
Für Geräte, die WLAN oder RFID nutzen, gilt die Richtlinie 2014/53/EU (Funkanlagen-Richtlinie bzw. Radio Equipment Directive, RED). Auch Medizinprodukte, die „funken“, etwa weil sie sich mit dem Internet verbinden oder über eine Fernbedienung angesteuert werden, müssen die Konformität mit der RED nachweisen, bevor sie auf den Markt dürfen.
Erfahren Sie in diesem Beitrag,
- welche Produkte unter die Funkanlagen-Richtlinie fallen,
- welche Anforderungen sie erfüllen müssen und
- wie Sie ohne unnötigen Prüfaufwand durch das Konformitätsbewertungsverfahren kommen.

1. Anwendbarkeit der RED
a) Was allgemein als Funkanlage gilt
Die RED definiert den Begriff “Funkanlage” in Artikel 2 Abs. 1 Nr. 1:
„ein elektrisches oder elektronisches Erzeugnis, das zum Zweck der Funkkommunikation und/oder der Funkortung bestimmungsgemäß Funkwellen ausstrahlt und/oder empfängt, oder ein elektrisches oder elektronisches Erzeugnis, das Zubehör, etwa eine Antenne, benötigt, damit es zum Zweck der Funkkommunikation und/oder der Funkortung bestimmungsgemäß Funkwellen ausstrahlen und/oder empfangen kann;“
Quelle: Artikel 2 Abs. 1 Nr. 1 RED
In Artikel 2 RED finden sich genauere Kriterien, wann eine Funkanlage unter die Richtlinie fällt und wann nicht. Dies hängt zum Beispiel von der Frequenz des Signals ab.
Vernetzte Geräte sind insbesondere von der RED betroffen, wenn sie WLAN, Zigbee, Bluetooth, RFID oder 4G/LTE/5G nutzen. Auch Systeme zu Abstandsmessungen wie Radare fallen in den Bereich der RED.
Beispiele für Produkte, die unter die RED fallen, sind unter Punkt 1c) aufgeführt.
b) Auf welche Produkte die RED nicht anwendbar ist
Es gibt zwei Gründe dafür, dass Funkanlagen nicht unter die RED fallen:
- Sie sind nicht vom Anwendungsbereich betroffen.
- Sie werden explizit von der RED ausgenommen.
Artikel 1 Abs. 3 und Anhang I der RED schließen bestimmte Funkanlagen ausdrücklich vom Anwendungsbereich der Richtlinie aus. Dazu zählen Funkanlagen zur Verteidigung und solche, die die öffentliche Sicherheit betreffen, Funkanlagen für Schiffe, die unter die Konventionen der International Maritime Organisation (IMO) fallen, und Amateurfunkgeräte.
Diese Ausnahmen betreffen also nicht explizit Medizinprodukte.
c) Medizinprodukte als Funkanlagen
Für Medizinprodukte gelten in erster Linie die Vorschriften der MDR und IVDR. Die RED gilt zusätzlich. Dies bedeutet: Die RED ist dann auf Medizinprodukte anwendbar, wenn diese Komponenten beinhalten, die unter die Definition der Funkanlage der RED fallen, und für die es in der MDR/IVDR keine speziellen Regelungen gibt. Beispiele für solche Komponenten sind WLAN- oder Bluetooth-Module.
Das gilt für alle Medizinprodukte, die bestimmungsgemäß mittels Funkwellen kommunizieren, unabhängig davon, ob sie direkt miteinander kommunizieren oder über andere IT-Geräte.
RED-Konformität
Betroffene Hersteller müssen zusätzlich zur MDR/IVDR auch die Konformität zur RED erklären. Kann der Nachweis der Konformität zur RED vollständig mit harmonisierten Normen erbracht werden, kann der Hersteller eine schriftliche EU-Konformitätserklärung ohne Hinzuziehen einer Benannten Stelle ausstellen. Andernfalls muss eine Benannte Stelle konsultiert werden. Für Frequenzbänder, die zum Beispiel von WLAN, BT oder RFID genutzt werden, sind harmonisierte Normen verfügbar.
Interessant ist, dass sich die Normen nicht auf eine spezifische Technologie beziehen, sondern nur auf Frequenzbänder. Innerhalb der Frequenzbänder können verschiedene Technologien entwickelt werden. So ist zum Beispiel die Norm ETSI EN 300 328 “Wideband transmission systems; Data transmission equipment operating in the 2,4 GHz band” für Technologien wie WLAN, BT oder Zigbee maßgebend.
Beispiele für Medizinprodukte, die unter die RED fallen
- Implantate, deren Funktion mit einer App kontrolliert werden kann
- Mobile Patientenmonitore, die Daten mit GSM übertragen
- Telemedizinische Geräte in der häuslichen Umgebung, die mittels WLAN Daten über das Internet übertragen
- Hörimplantate, die untereinander kommunizieren
- Produkte mit Smart Wireless Charging, die beim Laden Daten mit der Ladeeinheit austauschen
Beispiele für Medizinprodukte, die nicht unter die RED fallen
- Produkte, die Funkwellen nicht bestimmungsgemäß aussenden
- Medizinprodukte, die Funkstrahlung für therapeutische bzw. diagnostische Zwecke nutzen, z. B. Magnetfeldstimulation oder MRI
- Produkte, die nur drahtgebunden kommunizieren, z. B. mittels LAN, RS232 oder CAN Bus
RED, Cybersicherheit und Medizinprodukte
Mit der Delegierten Verordnung (EU) 2022/30 vom 29. Oktober 2021 gab es einige wesentliche Änderungen im Hinblick auf Cybersicherheit. Von diesen Regelungen sind Medizinprodukte, die unter MDR und IVDR fallen, laut Art. 2 Abs. 1 der Delegierten VO 2022/30 jedoch ausgenommen.
Es gelten jedoch andere Regelungen zum Thema Cybersicherheit für Medizinprodukte. Diese finden sich in der MDR sowie in der IEC 81001-5-1.
2. Vorgaben der RED
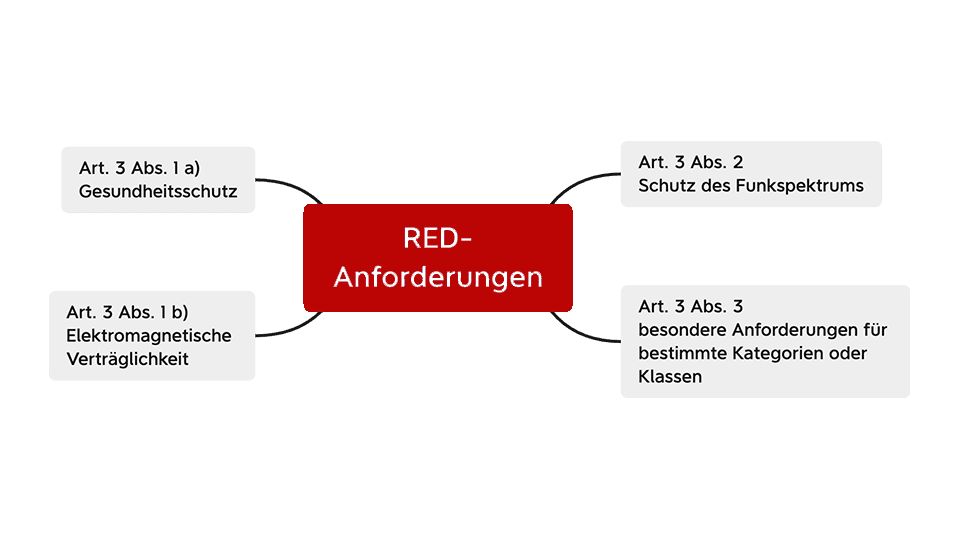
Die grundlegenden Anforderungen an Funkanlagen finden sich im Artikel 3 der RED.
Die wichtigsten Anforderungen sind:
- Art. 3 Abs. 1 a): Schutz der Gesundheit und Sicherheit von Menschen, Haus- und Nutztieren
- Art. 3 Abs. 1 b): Ein angemessenes Niveau an elektromagnetischer Verträglichkeit gemäß der Richtlinie 2014/30/EU
- Art. 3 Abs. 2: Funkanlagen müssen so gebaut sein, dass sowohl eine effektive Nutzung von Funkfrequenzen als auch eine Unterstützung zur effizienten Nutzung von Funkfrequenzen gegeben ist, damit keine funktechnischen Störungen auftreten.
- Art. 3 Abs. 3: Weitere konstruktive Anforderungen für bestimmte Kategorien oder Klassen, z. B.:
- 3(3)(d): Schutz des Netzes
- 3(3)(e): Sicherheitsvorrichtungen personenbezogener Daten und der Privatsphäre
- 3(3)(f): Schutz vor Betrug
Während MDR und IVDR spezifische Anforderungen im Anhang 1 formulieren, bleibt die RED vergleichsweise oberflächlich. Mehr Hintergründe zu den Gedanken und Erwägungen zu den Anforderungen finden sich im Guide to the Radio Equipment Directive 2014/53/EU (RED Guide).
a) Artikel 3 Abs. 1 a): Gesundheitsschutz
Hersteller müssen im Risikomanagement die biologischen Gefährdungen (Schutz der Gesundheit) im Zusammenhang mit nicht ionisierender Strahlung betrachten. Das ist insbesondere relevant für Implantate und Medizinprodukte, die am Körper getragen werden.
Die MDR formuliert im Anhang 1 Absatz 16 ebenfalls entsprechende Anforderungen zum Schutz vor Strahlung. Diese sind aber durch die Norm EN 60601-1 oder EN 60601-1-2 nicht vollständig abgedeckt. Hier helfen entsprechende Empfehlungen und Normen aus dem Umfeld der RED (Beispiele siehe Abschnitt 2c).
Referenzgrenzwerte und Basisgrenzwerte
Zur Festlegung der Vertretbarkeit der Risiken werden sogenannte Referenzgrenzwerte (ICNIRP) und Basisgrenzwert (SAR) festgelegt. Die Referenzgrenzwerte werden zum Beispiel für Implantate genutzt. Relevant sind Faktoren wie Leistungsdichte und Stromdichte, die zu einer Erwärmung des Gewebes führen. Auf der Webseite der Internationalen Strahlenschutzkommission ICNIRPS und im Guidance-Dokument ERC Recommendation finden Hersteller Empfehlungen und Normen. Zum Beispiel behandelt die ERC Recommendation im Anhang 12 Frequenzbänder und regulatorische sowie informative Parameter, die für aktive medizinische Implantate und ihre zugehörigen Peripheriegeräte empfohlen werden.
Die Internationale Strahlenschutzkommission sammelt Erkenntnisse über die biologische Wirkungsschwelle für hochfrequente elektromagnetische Felder (HF) im Bereich von 100 kHz bis 300 GHz und legt gewisse Grenzwerte fest.
b) Artikel 3 Abs. 1 b): Elektromagnetische Verträglichkeit
Artikel 3 Abs. 1 b) legt Anforderungen an die elektromagnetische Verträglichkeit fest. Ein Teil dieser Anforderungen ist bereits in der MDR und mit der Prüfung nach IEC 60601-1-2 für ME-Geräte abgedeckt.
Bezüglich der Funkeinrichtung werden zusätzlich Prüfungen der Störaussendung und der Immunität durchgeführt. Die Akzeptanzkriterien ähneln denen der IEC 60601-1-2 bezüglich der wesentlichen Leistungsmerkmale. Die Prüfungen sind in der Grundnorm EN 301 489-1 und ihrem Teil 17 EN 301 489-17 für BT und WLAN beschrieben.
c) Artikel 3 Abs. 2: Schutz des Funkspektrums
Funkeinrichtungen dürfen andere Bänder, insbesondere lizenzpflichtige Bänder wie Polizeifunk oder Militäranlagen, nicht übermäßig stören.
Es gibt jedoch per se keinen Ausschluss für die Wahl einer Technologie. Limits der Technologie ergeben sich aus dem Anwendungsfall (intended environment) und den darin geltenden Bestimmungen: Military Standard (MIL), Radio Technical Commission for Aeronautics (RTCA), European Telecommunications Standards Institute (ETSI).
Innerhalb der Bänder gibt es international starke Unterschiede, da die Funkfrequenzen in der Hoheit der Staaten liegen. Das Frequenzband 2,4 GHz, in das auch WLAN und BT fallen, ist eines der wenigen Frequenzbänder, das im größten Teil der Welt lizenzfrei ist.
Die wichtigsten Normen für ausgewählte Frequenzbänder
- EN 300 328 2.4GHz BT, WLAN, Zigbee
- EN 301 893 WLAN, 5G Hz
- EN 300 330 RFID 13.56 MHz
Die Normen beziehen sich nie auf eine bestimmte Anwendung wie WLAN oder RFID, sondern nur auf Frequenzbereiche und Übertragungstechnologien. Das hat den Vorteil, dass weitere Funkanwendungen entwickelt werden können, ohne dass neue Normen notwendig sind.
d) Artikel 3 Abs. 3: Besondere Anforderungen
Artikel 3 Abs. 3 legt besondere Anforderungen für “bestimmte Kategorien oder Klassen” fest. Diese Anforderungen beziehen sich vor allem auf die Themen Kompatibilität und Sicherheit. Welche Klassen und welche Kategorien dies genau betrifft, findet sich in den Delegierten Rechtsakten zur RED.
Die Artikel 3 Absatz 3 Buchstabe d, e und f legen Anforderungen fest, die sich auch auf Elemente der Cybersicherheit beziehen und auf jene Kategorien von Funkanlagen anwendbar sind, die Risiken für die Cybersicherheit darstellen. Diese gelten jedoch nach Art. 2 Abs. 1 der Delegierten Verordnung (EU) 2022/30 nicht für Medizinprodukte nach MDR und IVDR.
MDR und IVDR haben jedoch eigene Vorgaben zum Thema Cybersecurity.
Eine Übersicht über alle wichtigen Dokumente inklusive Guidance-Dokumente gibt es auf den Seiten der EU zur RED.
3. Einbau von Funkmodulen in Medizinprodukte
Hersteller bestücken ihre Medizinprodukte meist mit bereits existierenden Funkmodulen und entwerfen diese nicht neu. Diese Module sind bereits RED-konform und besitzen eine entsprechende Bescheinigung. Dies bedeutet jedoch nicht, dass das Medizinprodukt automatisch RED-konform ist.
Werden bereits bestehende Funkmodule in einem Medizinprodukt verbaut, muss in den allermeisten Fällen für das gesamte Produkt erneut die RED-Konformität nachgewiesen werden.
Dies liegt daran, dass sich die Eigenschaften durch den Einbau meistens verändern. Es entsteht also ein neues Funkgerät, das wiederum die Anforderungen der RED erfüllen muss.
Funkmodule haben im eingebauten Zustand einen veränderten EMV-Fingerabdruck. Das betrifft die Topologie der Antenne, die Ansteuerung mittels Software und die Qualität der Spannungsversorgung. In der EU ist daher ein Test im verbauten Zustand gefordert.
a) Wann Hersteller dennoch auf gewisse Prüfungen verzichten können
Unter Umständen können Hersteller jedoch auf einen Teil der Prüfungen für die RED verzichten, wenn sie in ihrem Medizinprodukt bereits ein RED-konformes Funkmodul verwenden. Die Entscheidung sollte in der Risikoanalyse getroffen werden.
Als Daumenregeln lassen sich festhalten:
- Alle Elemente, die geleitet (“conducted”) angeschlossen sind, verändern sich durch den Einbau i. d. R. nicht. Sie müssen daher nicht zwangsläufig erneut geprüft werden.
- Elemente, die strahlen (“radiated”), müssen nach EMV sowie Art. 3 Abs. 2 RED i. d. R. komplett neu geprüft werden.
- Werden mehrere Funkgeräte verbaut, macht deren Zusammenwirken meist eine Prüfung nötig.
- Bei Modulen, die außenliegend installiert sind, kann oft auf eine erneute Prüfung verzichtet werden.
- Module, die tief verbaut sind, müssen meist erneut geprüft werden.
Welche Teile der Prüfung erneut für das Medizinprodukt durchgeführt werden müssen, unterliegt jeweils einer Einzelfallentscheidung. Ändern sich die Eigenschaften des Moduls durch den Einbau, können schwere Schäden entstehen!
Oft sind zumindest Teilprüfungen nötig, wie die Nebenaussendungen (Spurious Emissions) nach dem jeweiligen Funkstandard. Für die genaue Einschätzung ist Zugriff auf den Prüfbericht des verbauten Moduls hilfreich.
b) Tipps zum Einbau von Funkmodulen
- Tipp 1: Achten Sie bei Medizinprodukten auf die Qualität des verwendeten Funkmoduls. Dadurch stellen Sie nicht nur die Sicherheit und Leistung sicher, sondern können sich auch viele Probleme beim Support ersparen.
- Tipp 2: Medizinprodukte haben eine längere Lebensdauer als andere Funkgeräte wie Smartphones. Achten Sie daher auf eine gute Dokumentation und eine lange Servicezeit beim verwendeten Funkmodul.
- Tipp 3: Versuchen Sie mit den Antennen auszukommen, die in der Installationsanweisung definiert sind. Sollten Sie Änderungen vornehmen, ist der Konformitätsnachweis hinfällig!
- Tipp 4: Achten Sie vor dem Einbau darauf, was zu welchen neuen Prüfungen führt. Stellen Sie in der Risikoanalyse dezidiert fest, was Sie wirklich noch einmal prüfen müssen und was nicht. Wenn Sie einen guten Plan aufstellen, können Sie sich unter Umständen aufwändige Doppelprüfungen sparen.
4. In fünf Schritten zur RED-Konformität
Damit Sie Ihr Medizinprodukt mit Funkmodul sicher durch das Konformitätsbewertungsverfahren zur RED bringen, sollten Sie die folgenden Punkte im Auge behalten:
a) Schritt 1: Ziel festlegen
Überlegen Sie sich zunächst genau, was Sie mit welcher Bandbreite wie weit und mit welchen weiteren Anforderungen übertragen wollen. Legen Sie danach den geeigneten Frequenzbereich fest. Zu den Überlegungen gehören Aspekte wie Datendurchsatz, Reichweite, Verfügbarkeit oder Sicherheit. Der Ansatz „so viel wie nötig und so wenig wie möglich“ sollte im Vordergrund stehen.
b) Schritt 2: Anwendbarkeit prüfen
Prüfen Sie, ob Ihr Produkt überhaupt in den Anwendungsbereich der RED fällt. Die Voraussetzungen finden Sie in Art. 2 RED (vgl. auch oben Punkt 1a).
c) Schritt 3: Anforderungen feststellen
Stellen Sie fest, welche Anforderungen Ihr Produkt nach Art. 3 RED zusätzlich zu den Anforderungen der MDR erfüllen muss. Am besten erfassen Sie diese zusammen mit den Anforderungen anderer Regelungen, die für Ihr Produkt einschlägig sind, etwa Regelungen der MDR oder IVDR.
Beachten Sie: Zwar sind Medizinprodukte von den Anforderungen bezüglich Cybersecurity der RED ausgenommen, dennoch sollten Sie auf diese großen Wert legen. Cybersecurity ist nach anderen Regelungen wie der MDR für Medizinprodukte verpflichtend.
d) Schritt 4: Funkmodul bei Risikoanalyse in den Fokus nehmen
Legen Sie bei der Risikoanalyse einen besonderen Fokus auf das Funkmodul. Hierbei können Sie frühzeitig feststellen, welches Funkmodul Sie wie einbauen und sich damit später unnötige Prüfungen sparen. Beachten Sie dabei auch die Besonderheit beim Einbau (vgl. Punkt 3 oben).
e) Schritt 5: Auf RED-Konformität prüfen
Die Anhänge der RED liefern genauere Hinweise zur Prüfung auf RED-Konformität. Die Normenreihen EN 301 489 und EN 60601-1-2 kommen dabei meist zur Anwendung. Prüflabore unterstützen Sie bei der Festlegung der notwendigen Prüfungen.
5. Aktuelles
a) Änderung der RED-Richtlinie
Die EU hat eine Änderung zur RED-Richtlinie (Funkanlagen-Richtlinie) veröffentlicht. Die RED betrifft auch Medizinprodukte, die kabellos kommunizieren, z. B. via Bluetooth oder WLAN.
„Funkanlagen“ müssen einen einheitlichen Ladeanschluss haben, nämlich USB-C. Die Gesetzgeber hatten damit v. a. Mobiltelefone im Fokus, Medizinprodukte sind jedoch nicht ausgenommen.
Hersteller sollten daher überprüfen, ob ein zusätzlicher Patientenschutz im Medizinprodukt selbst eingebaut werden muss, falls nicht sichergestellt werden kann, dass die Anwender immer das mitgelieferte Netzteil verwenden.
b) Auswirkungen der Richtlinie 2022/2380 („USB“)
Anforderungen
Die Vorschrift, dass Funkanlagen mit dem einheitlichen Ladeanschluss USB Typ C ausgestattet sein müssen, greift ab dem 28. Dezember 2024 für bestimmte Kategorien oder Klassen von Funkanlagen. Das fordert die Richtlinie (EU) 2022/2380 im Anhang Ia Teil I Nummern 1.1 bis 1.12). Der Deutsche Gesetzentwurf zur Umsetzung der Richtlinie (EU) 2022/2380 liegt vor.
Anwendungsbereich
Betroffen sind beispielsweise folgende Kategorien (Funkanlagen)
- tragbare Mobiltelefone
- Tablets
- Headsets
- Ohrhörer
Für Laptops sind diese Vorschriften ab 28. April 2026 gültig.
Konsequenzen für die Geräte
Wenn solche Komponenten vom Patienten genutzt werden müssen, damit das Medizinprodukt seine klinische Funktion erfüllen kann, gelten höhere Sicherheitsanforderungen für diese Komponenten an den Patientenschutz. Ein Problem kann auftreten, wenn der Patient das Gerät (zum Beispiel einen Laptop) verwendet, während es mit dem Netzteil verbunden ist.
Bisher haben Hersteller ein Ladegerät verwendet, das den Patientenschutzanforderungen entspricht und einen eigenen Anschlussstecker bereitstellt. Dies ist nun nicht mehr möglich. Jetzt müssen sie überprüfen, ob das verwendete USB-C-Netzteil die Anforderungen der IEC 60601-1 erfüllt, oder sicherstellen, dass Anwendung und Aufladen nicht gleichzeitig möglich sind.
6. Fazit
Mit zunehmender Vernetzung von Medizinprodukten fallen diese immer häufiger unter die Funkanlagen-Richtlinie RED. Sie als Hersteller sollten sich daher unbedingt mit den Anforderungen vertraut machen. Ein besonderes Augenmerk müssen Sie dabei auf die Risikoanalyse legen. Das Einbauen bereits zertifizierter Funkmodule kann neue Risiken und hohe Kosten mit sich bringen, wenn vorab nicht alle Aspekte bedacht werden. Bei Fragen können Sie sich an Prüfstellen oder das Johner Institut wenden.
Wenn Sie Fragen zur RED haben oder Hilfe beim Konformitätsbewertungsverfahren benötigen, nehmen Sie gern Kontakt zu den Expertinnen und Experten des Johner Instituts auf.
Wir bedanken uns herzlich bei eurofins für wertvollen Input zu diesem Beitrag.
Änderungshistorie
- 2024-02-02: Teilkapitel 5. b eingefügt
- 2023-02-03: Teilkapitel „Änderung der RED-Richtlinie“ ergänzt


Die RED-Richtlinie ist komplexe Materie, für welche viele Medizinprodukte-Hersteller nicht oder nur begrenztes Know-How haben. Allerdings ist aufgrund der Ausnahme-Klauseln oftmals keine tiefe Detail-Kenntnis nötig. Es wäre daher schön und hilfreich, wenn etwas genauer auf diese Aufnahmen-Klauseln eingegangen werdne könnte.
Konkret stellt sich die Frage, wie es sich mit RFID-Tags verhält:
– viele Medizinprodukte-Hersteller kaufen fertige Tags ein, programmieren diese, und bringen sie als Komponenten eines Medizinprodukts an
– RFID-Tags interagieren immer mit Komponenten anderer Systeme, d.h. mit einem RFID-Schreib- und/oder -Lesekopf. Diese Schreib-/Leseköpe sind teilweise in anderen Geräten oder gar Medizinprodukten verbaut, entsprechend sind mehrere Geräte involiert.
Daher ergeben sich folgende Fragen:
– müssen Medizinprodukte, welche einen handelsüblichen RFID-Tag enthalten, ebenfalls geprüft werden?
– muss für eine solche Prüfung das Gerät, welches den Lese-/Schreibkopf enthält, in der Prüfung auch berücksichtigt werden, wenn dieser in einem anderen Gerät bzw. Medizinprodukt verbaut ist?
Wäre super, wenn dieser oder ähnliche Fälle etwas auführlicher bzw. konkreter beschrieben werden könnten.
Lieber Herr Reichmuth,
genau auf die Problematik der Nahfeldkommunikation geht die IEC 60601-1-2 im Amendment 1 ein und verlangt für Produkte mit magnetisch sensitiven Teilen eine zusätzliche Prüfung im Bereich der Störfestigkeit nach der IEC 61000-4-39. Die früheren Warnhinweise Geräte nicht zu stapeln oder einen Abstand von 15cm zu lassen, betrachtet die IEC 60601-1-2 nicht mehr als angemessen bzw. Stand der Technik. Die Intention der Prüfung kommt daher aus der EMV Norm und nicht aus der RED.
Liebe Grüsse, Mario Klessascheck
Hallo Herr Klessacheck,
bei der Verwendung von einem zugelassenem Funk-USB-Stick stellt sich mir die Frage, was „außenliegend installiert“ definiert: muss der USB-Steckplatz von aussen direkt frei und sichtbar sein? Kann das ein Fach mit Klappe (ähnlich typisches Batteriefach) sein? Kann man als Grundregel verwenden: „der Anwender muss den Funk-USB-Stick ohne Werkzeug und ohne großen Aufwand selbst installieren können“?
Gruß
R. Wagner
Lieber Herr Wagner,
Man unterscheidet dabei zwischen fest verbaut und nicht fest verbaut. Einen USB Stick würde ich daher als nicht fest verbaut betrachten, auch nicht, wenn er sich hinter einer Schutzabdeckung befindet.
beste Grüsse, Mario Klessascheck
Dann ist der Begriff „aussenliegend“ aber völlig ad absurdum geführt. Es ist technisch ohne weitres möglich, eine USB-Buchse im tiefsten Inneren eines Geräts zu verbauen, sodass nur der Techniker nach Demontage etlicher Teile ran kommt. Das ist alles andere als aussenliegend und trotzdem nicht fest verbaut. Das wird bei vielen Geräten heute üblich.
Hallo Herr Klessacheck,
ist es denn sicher, dass auch Medizinprodukte von der Umstellung auf USB-C-Ladebuchsen betroffen sind? Und ab wann würde das gelten? Die endgültige Verabschiedung ist ja noch nicht erfolgt.
Hintergrund: Ich arbeite für einen Kunden, der gerade mit der MDD-Zulassung für ein neues Produkt Klasse IIa anfängt. Das Produkt beinhaltet ein BT-Modul und eine herkömmliche Ladebuchse. Da nicht absehbar ist, ob der Markteintritt vor Mitte 2024 erfolgt, stellt sich die Frage, ob wir während des laufenden Verfahrens jetzt eine Designänderung vornehmen müssen.
Lieber Herr Völpel,
Es ist nicht sicher, dass Medizinprodukte davon betroffen sind. Im Anhang Ia der Richtlinie werden Kategorien und Klassen genannt, die in den Geltungsbereich fallen. Wenn ihr Produkt nicht in eine dieser Kategorien fällt, dann wäre das Produkt generell nicht betroffen. Das Sie die Frage stellen und den Aspekt in der Produktdokumentation bewerten, wollten wir mit dem Artikel erreichen.
Liebe Grüsse, Mario Klessascheck
Guten Tag,
ich habe da eine Frage:
Im neuen Journal steht folgende Anmerkung:
„Funkanlagen“ müssen einen einheitlichen Ladeanschluss haben, nämlich USB-C. Der Gesetzgeber hatte bei der Änderung zwar Mobiltelefone im Fokus, Medizinprodukte sind jedoch nicht ausgenommen“.
Das würde bedeuten, dass man seinem Medizinprodukt nun einen USB-C- Anschluss spendieren muss und sich gleichzeitig noch um die Patientensicherheit kümmern sollte.
Ich frage mich nur, wie und warum ich eine 4kV-sichere Trennung im Gerät realisieren soll, nur dass der Anwender das Gerät mit einem handelsüblichen USB-C Ladegerät aufladen kann, welches bei weitem nicht den Standards eines med. Netzteils entspricht.
Wo liegt hier die Sinnhaftigkeit?
Wie soll ein Hersteller sicherstellen, dass immer das mitgelieferte, Patientensichere Netzteil verwendet wird?
Besteht die Chance, dass dieser Punkt noch überarbeitet wird?
Lieber Herr Schlaich,
sie schlussfolgern völlig richtig. Die Richtlinie nennt im Anhang Ia Klassen von Produkten, die den Geltungsbereich fallen. Medizinprodukte werden nicht explizit erwähnt, sondern eher klassische Konsumerprodukte, wie Kameras oder Tablets. Es kann aber sein, dass solch ein Produkt auch ein Medizinprodukt wird. Wenn es kein Anwendungsteil gibt, dann ist es auch keine ME-Gerät, falls doch würde man gleichzeitiges Nutzen und Laden verhindern und dann erfüllt USB-C auch die Vorgaben der IEC 60601-1.
Vielen Dank für Ihre Gedanken.
Liebe Grüsse, Mario Klessascheck
Hallo Herr Klessascheck,
sie schrieben: „Wenn es kein Anwendungsteil gibt, dann ist es auch kein ME-Gerät…“
Wie haben Sie das konkret gemeint?
Das Vorhandensein eines Anwendungsteils ist noch keine Voraussetzung für ein ME-Gerät?
Viele Grüße
Lieber Herr Killian,
Der Geltungsbereich der IEC 60601-1 bezieht sich auf ME-Geräte. ME-Geräte sind in der Norm folgendermaßen definiert: „electrical equipment having an APPLIED PART or transferring energy to or from the PATIENT or detecting such energy transfer to or from the PATIENT.“ Das Anwendungsteil ist der Teil des Produktes, dass zwingend mit dem Patienten in Kontakt kommen muss. Der Begriff ME-Geräte bezieht auch Produkte ein, die keinen Kontakt haben, sondern bei denen eine Energieübertragung stattfindet, zum Bsp. Röntgengeräte. Somit sind das Anwendungsteil oder die Energieübertragung eine Bedingung für den Geltungsbereich.
Liebe Grüsse, Mario Klessascheck
Im betreffenden Anhang 1a der Änderung der RED-Richtlinie sind keine Medizinprodukte aufgeführt.
Lieber Herr Schmid,
Das stimmt. Es sind allerdings Produkte aufgeführt, die wir auch schon als Medizinprodukte gesehen haben. Ich hatte beim Lesen keinen Hinweis auf den Ausschluss von Medizinprodukten gefunden und wollte erst einmal ein Achtung signalisieren.
Liebe Grüsse, Mario Klessascheck
Wenn man weiter gräbt, dann kommt heraus, dass die USB-C-Buchse zu Aufladen der Batterie im Sinne von RED eben NICHT für Medizinprodukte gilt.
ACHTUNG! Die aktuelle konsolidierte Fassung der RED und das aktuelle Amendment weichen ab!!!
Im Amendment (DIRECTIVE (EU) 2022/2380) werden nämlich die betroffeneren Geräte aufgezählt, Medizingeräte sind nicht dabei:
https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:32022L2380&from=EN#d1e34-40-1
1.1. handheld mobile phones; 1.2. tablets; 1.3. digital cameras; 1.4. headphones; 1.5. headsets; 1.6. handheld videogame consoles; 1.7. portable speakers; 1.8. e-readers; 1.9. keyboards; 1.10. mice; 1.11. portable navigation systems; 1.12. earbuds; 1.13. laptops.
Diese Aufzählung fehlt in der konsolidierten Fassung der RED.
https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:02014L0053-20221227&qid=1675785103580&from=en#tocId65
Lieber Herr Steinke,
vielen Dank für Ihren wertvollen Hinweis und das Sie das mit uns teilen. Sie haben recht, Medizinprodukte werden nicht erwähnt, aber auch nicht ausgeschlossen. Es kann nun sein, dass ein Tablet oder eine Kamera ein Medizinprodukt ist und daher kam unser Zeigefinger. Hersteller sollten diese Änderung im PMS Prozess bewerten und einen Ausschluss begründen.
Liebe Grüsse, Mario Klessascheck
Hallo Herr Klessacheck,
wie ist es denn bei Medizinprodukten, die nicht implantiert werden, aber gemessene Werte an eine App per Bluetooth übertragen?
Viele Grüße
Lisa Schneider
Liebe Frau Schneider,
Der Geräteteil mit Bluetooth fällt in den Anwendungsbereich der RED.
Liebe Grüsse, Mario Klessascheck
Hallo Herr Klessacheck,
das Thema USB C Ladestecker beschäftigt uns ebenfalls sehr. Unser ME-Gerät misst Druck und verfügt über Funktechnologie um diese Druckwerte zu übertragen, somit fällt es in den Anwendungsbereich der RED. Wir liefern dazu eine Art Ladeschale, welche über zwei Kontakte unser ME-Gerät lädt und selbst mittels eines einfachen USB Steckernetzteil versorgt wird.
1) Müssen wir das Prinzip des Ladens unseres ME-Geräts über zwei Kontakte, durch ein USBC Stecker ersetzen?
2) Oder sollte nun die Ladeschale einen USB C Anschluss bekommen, obwohl diese nicht über eine Funktechnologie verfügt?
Viele Grüße,
Dirk Brosius
Lieber Herr Brosius,
Die Spezifikationen in Anhang Ia Teil I der Richtlinie 2014/53/EU (geändert in der Richtlinie (EU)2022/2380) betrifft nur bestimmte Kategorien wie: Tablets, Tastaturen, Digitalkameras etc. Ich vermute, dass Ihr Druckmessgerät nicht in eine dieser Kategorien fällt. Daher müssen Sie die Anforderung nicht umsetzen.
Liebe Grüße, Mario Klessascheck
Hallo Herr Klessascheck,
vielen Dank für den hilfreichen Beitrag und die Aktualisierungen zum Thema Funk. Können Sie uns sagen, ob es zulässig ist, für unser Medizinprodukt, welches ein WiFi-Modul integriert hat, 2 einzelne Konformitätserklärungen (eine für MDR, eine für RED) zu erstellen? Für unser Ablagesystem wäre dies einfach übersichtlicher, wir sind uns aufgrund des RED Artikels 18(3)* jedoch nicht sicher, ob wir dies dürfen.
(* Unterliegt eine Funkanlage mehreren Rechtsakten der Union, die eine EU-Konformitätserklärung vorschreiben, wird für alle Rechtsakte der Union eine einzige EU-Konformitätserklärung ausgestellt. In dieser Erklärung sind die betroffenen Rechtsvorschriften der Union samt ihrer Fundstelle im Amtsblatt anzugeben.)
Vielen herzlichen Dank vorab für Ihre Antwort.
Beatrice Dachsel
Liebe Frau Dachsel,
Sie haben bereit die richtige Textstelle gefunden. Für ein Produkt können Sie in seiner Sachgesamtheit nur eine einzige Konformitätserklärung ausstellen. Sie könnten höchstens das Funkmodul als separates Modul betrachten und dann eine separate Konformitätserklärung ausstellen. Sie müssten in dem Fall das Funkmodul zusammen mit Ihrer Maschine bewerten und in der Konformitätserklärung den Bezug zu der Maschine herstellen (Bsp. Funkmodul für Maschine XY). Beachten Sie aber bitte, dass Sie dann auch eine Produktdokumentation erstellen müssen, wie sie in der RED gefordert ist.
Liebe Grüsse, Mario Klessascheck