
Mit dem 21 CFR part 822 legt die FDA die Anforderungen an die Post-Market Surveillance fest. Ein zugehöriges „Guidance Document“ gibt Handlungsleitung, wie Hersteller die Forderungen des 21 CFR part 822 erfüllen sollen.
21 CFR part 822 im Überblick
Der 21 CFR part 822 regelt,
- wann Hersteller eine Post-Market Surveillance (PMS) durchführen (lesen Sie hier einen ausführlichen Artikel zur Post-Market Surveillance),
- wie die Hersteller diese Marktüberwachung planen,
- welche Unterlagen sie mit welchen Inhalten erstellen und
- welche Fristen sie und die FDA einhalten müssen sowie
- wie die FDA die Konformität prüft.
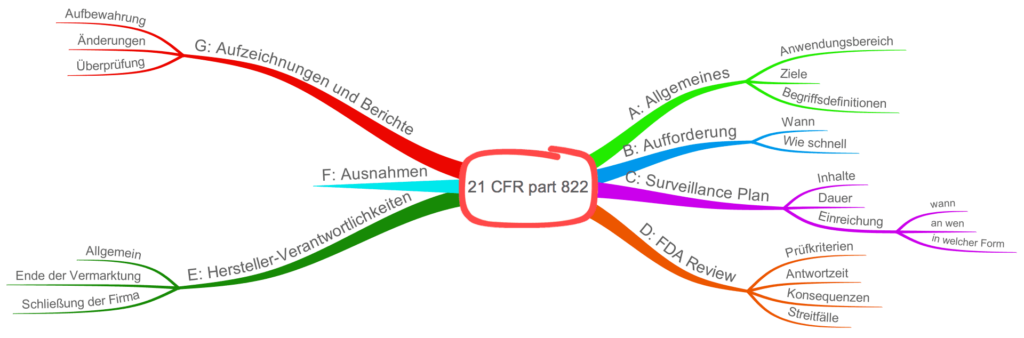
21 CFR part 822: Zum Vergrößern klicken
Der 21 CFR 822 weißt zumindest für europäische Leser Besonderheiten auf:
- Der 21 CFR part 822 liegt als FAQ vor: Die einzelnen „parts“ sind als Fragen mit zugehörigen Antworten formuliert. Beispielsweise ist der §822.1 übertitelt mit „What does this part cover?“.
- Hersteller müssen nur dann eine PMS durchführen, wenn sie von der FDA dazu aufgefordert werden
- Die Hersteller müssen den PMS-Plan von der FDA genehmigen lassen, bevor sie mit der Umsetzung beginnen.
- Selbst wenn der Hersteller das Produkt vom Markt nimmt, behält es sich die FDA vor, auf der Fortsetzung der PMS zu bestehen.
Wann Hersteller eine Post-Market Surveillance durchführen müssen
Hersteller müssen eine PMS durchführen, wenn die FDA sie dazu auffordert. Die FDA ist laut 21 CFR part 820.1 dazu befugt, wenn das Medizinprodukt
- In die Klasse II oder III fällt
- und ein Fehlverhalten zu schwerwiegenden Gesundheitsstörungen führen könnte
- oder das Gerät für mehr als ein Jahr implantiert werden darf
- oder das Gerät lebensherhaltend oder lebensunterstützend wirkt
Die FDA kann diese Forderung gleich beim Zulassungsantrag oder zu jedem anderen späteren Zeitpunkt erheben.
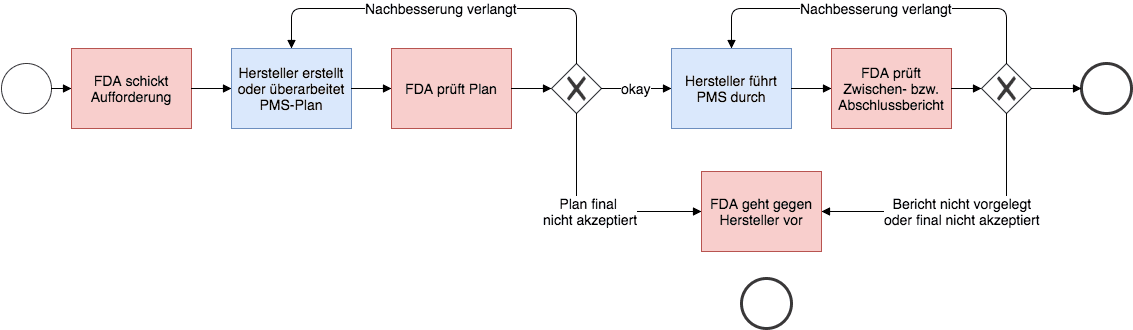
Fristen
Der 21 CFR part 822 nennt einige Fristen für Hersteller und die FDA selbst:
- Die Hersteller müssen 30 Tage nach Aufforderung den PMS-Plan einreichen.
- Die FDA lässt sich bis zu 60 Tage zum Review Zeit.
- Fristen zur Nachbesserung legt die FDA im Einzelfall fest.
- Änderungen bezüglich der Firma (Eigentümer, Schließung) müssen innerhalb 30 Tagen erfolgen.
- Aufzeichnungen müssen Hersteller mindestens 2 Jahre nach dem Zeitpunkt aufbewahren, an dem die FDA den Abschlussbericht genehmigt hat.
Inhalte
Zu den Inhalten, die die Hersteller bei Aufforderung zur Verfügung stellen müssen, zählen:
- Angaben zur Firma und zum Produkt (insbesondere zur Identifikation und Zulassung)
- Post-Market Surveillance Plan (dazu gleich mehr)
- Ansprechpartner
Der PMS-Plan muss enthalten:
- Ziele der Überwachung
- Studienobjekt (typischerweise Patienten oder das Produkt)
- Erfolgskriterien
- Vorgehen / Verfahren inklusive Datenquellen, Anzahl Probanden oder Geräte
- Überwachung der Studie,
- Dauer (die FDA kann bis zu 36 Monate verlangen),
- Auswertung und Berichtswesen
Konsequenzen
Wenn der Hersteller nicht in der Lage ist, einen Post-Market Surveillance Plan zu erstellen, dem die FDA zustimmt, oder diesen Plan umzusetzen, droht die FDA mit Konsequenzen wie Strafzahlungen oder gar Strafverfolgung. Sie kann „actions against products“ einleiten.
Aufzeichnungen
Die Hersteller müssen gemäß 21 CFR part 822.31 folgende Aufzeichnungen aufbewahren:
- Korrespondenz mit der FDA
- Vereinbarungen mit den „Investigators“ (z.B. Prüfärzten)
- Der von der FDA freigegebene PMS-Plan
- Alle Daten, die in diesem Kontext erhoben werden
- Weitere Unterlagen wie Einverständniserklärungen
Die FDA behält sich nicht nur vor, die Unterlagen vor Ort einzusehen, sondern auch Inspektionen an allen Orten durchzuführen, die an der PMS beteiligt waren.
Guidance Document
Die FDA hat ein Guidance Document mit dem Titel „Postmarket Surveillance Under Section 522 of the Federal Food, Drug, and Cosmetic Act“ veröffentlicht.
Dieses Guidance Document
- Wiederholt die regulatorischen Anforderungen
- Ergänzt die Liste von Inhalten eines PMS Plans um konkretere Beispiele
- Nennt Beispiele für Methoden einer PMS wie
- Retrospektive Kohortenstudie
- Aktive Marktüberwachung
- Tierversuche
- Laborversuche
- Meta-Analyse von anderen Studien
- Konkretisiert die Anforderungen an das Berichtswesen: die FDA verlangt typischerweise im Abstand von sechs Monaten Zwischenberichte
- schreibt vor, was die Zwischen- und Abschlussberichte enthalten müssen
- Listet die Status der Bearbeitung (das entspricht dem oben gezeigten Prozessdiagramm)
- Enthält die Checkliste, mit der FDA-Mitarbeiter den PMS-Plan prüfen.


Sehr geehrte Damen und Herren,
mir stellt sich in dem Zusammenhang die Frage, inwiefern sich die PMS und Vigilanz der FDA und der neuen Regelungen der MDR noch unterscheiden? Wurden die Anforderungen soweit angeglichen, dass ein Markteintritt für ein Medizinprodukt, das bereits in Europa zugelassen ist, die PMS Anforderungen soweit erfüllt hat, dass ein Markteintritt in den USA fast barrierefrei wäre?
Oder übersehe ich große Diskrepanzen zwischen den Anforderungen der CFR 21 820 und der MDR?
Vielen Dank im voraus,
Paul
Sehr geehrter Paul,
die PMS-Anforderungen sind v.a. Anforderungen an den Prozess, auch wenn diese produktspezifisch formuliert werden müssen.
Die Anforderungen an die Vigilanz unterscheiden sich, auch wenn sie sich ähneln. Die Vigilanz unterscheidet sich in Europa sogar von Land zu Land (noch). Die Meldefristen, die Form der Meldung, der Inhalt der Meldung, der Übermittlungsweg bzw. der Empfänger sind aber in der EU und den USA so unterschiedlich, dass es dazu spezifischer Arbeitsanweisungen bedarf.
Beste Grüße, Christian Johner