
Die FDA hat die Bedeutung der Interoperabilität von Medizinprodukten früh erkannt und 2017 das Guidance Dokument ‚Interoperable Medical Devices‘ veröffentlicht.
Die US-Behörde möchte damit der Tatsache Rechnung tragen, dass die Interoperabilität von Medizinprodukten einerseits wichtig für die Gesundheitsversorgung ist. Andererseits führen Probleme mit mangelnder Interoperabilität zu Risiken.
Dieser Beitrag verschafft Ihnen einen schnellen Überblick über die Anforderungen der FDA an ‚Interoperable Medical Devices‘ und gibt Tipps wie Sie diese erfüllen können.
Ein eigener Artikel zur Interoperabilität führt in das Thema ein und nennt die gesetzlichen Anforderungen, die in anderen Rechtsbereichen als den USA gelten.
1. Über das Guidance Document
a. Wen das FDA Dokument betrifft
Das Guidance Document ‚Interoperable Medical Devices‘ wendet sich an Hersteller, die Medizinprodukte mit Datenschnittstellen in den USA verkaufen wollen. Die Behörde formuliert darin ihre Erwartung an die „Zulassungsunterlagen“.
Zudem möchte die FDAmit diesem Dokument gleichzeitig auch den eigenen Inspektoren das Verständnis der eigenen Behörde zu diesem Thema vermitteln.
Wie alle „Guidance Documents“ ist auch dieses nicht „legally binding“. Dennoch erwartet die FDA, dass die Hersteller bei Einreichungen (z.B. alle Formen der 510(k), PMA) die in diesem Dokument geforderten Informationen beilegen!
Zudem sind Hersteller, die in die USA importieren, generell gut beraten, alle FDA „Guidance Documents“ zu befolgen.
Gleichzeitig liefert dieses Dokument auch Medizinprodukteherstellern außerhalb des US-Markts wertvolle Hinweise zum Entwickeln interoperabler Medizinprodukte.
b. Was die FDA unter Interoperabilität versteht
Die FDA folgt den üblichen Definitionen des Begriffs „Interoperabilität“. Demnach versteht man unter Interoperabilität die Fähigkeit von zwei oder mehr Produkten, Technologien oder Systemen, Informationen auszutauschen und ausgetauschte Informationen zu nutzen.
Lesen Sie in diesem Artikel mehr darüber, was man unter Interoperabilität versteht und welche Interoperabilitätsebenen man unterscheidet.
2. Anforderungen an ‚Interoperable Medical Devices‘
Das mit 18 Seiten erfreulich kurze Dokument führt in den ersten vier Kapiteln ins Thema ein, und benennt den Anwendungsbereich sowie Definitionen.
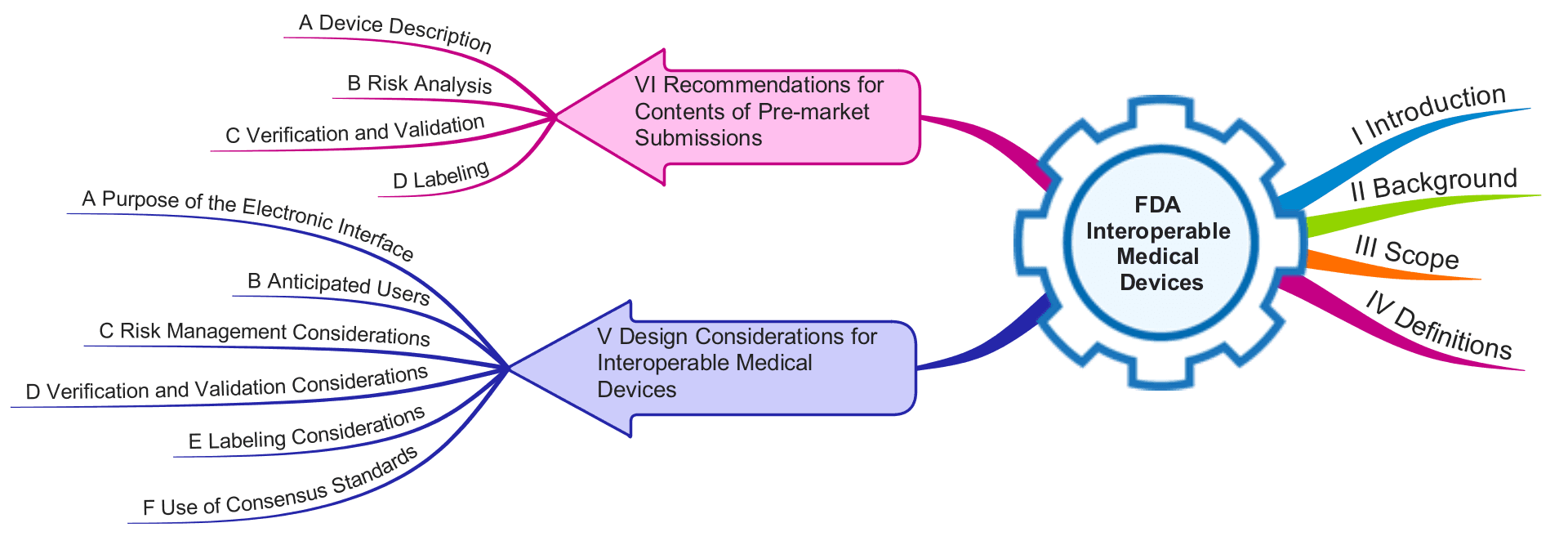
Die wesentlichen Forderungen finden sich in den Kapiteln V und VI:
- Das Kapitel V gibt Empfehlungen, was man beim Entwickeln interoperabler Medizinprodukte beachten soll.
- Das Kapitel VI legt fest, welche Informationen die FDA bei Einreichungen erwartet.
Kapitel V: ‚Design Considerations for Interoperable Medical Devices‘
Die FDA verlangt, dass die Hersteller beim Entwurf interoperabler Medizinprodukte die folgenden Aspekte adressieren:
A) Zweck der (elektronischen) Schnittstelle
Die Hersteller müssen in einer Zweckbestimmung festlegen,
- welche Daten
- in welchem Format,
- in welcher Weise (Protokolle, uni-/bidirektional etc.)
- zwischen welchen Geräten
- zu welchem Zweck (z.B. Alarm übertragen, Gerät ansteuern)
- in welchem klinischen Kontext (z.B. bei Beatmung von Patienten)
ausgetauscht werden sollen.
Damit fordert die FDA nicht nur den „Zweck“ („purpose“) zu beschreiben, sondern die Schnittstelle zu spezifizieren.
B) Vorgesehene Anwender
Die FDA nennt Anwendergruppen wie Kliniker, Service-Personal, Systemintegratoren, Mitarbeiter der IT und Patienten. Diese verfolgen bei der Nutzung der Schnittstelle verschiedene Ziele und verfügen über unterschiedliches Wissen.
Hersteller müssen diese Anwendergruppen kennen und beim Risikomanagement berücksichtigen.
C) Risikomanagement
Die Hersteller sind verpflichtet, die Risiken durch mangelnde Interoperabilität zu identifizieren und zu beherrschen. Dazu zählen Risiken durch falsche Daten oder Kommandos, durch Fehlfunktionen der Schnittstellen, durch Probleme mit der IT-Security oder dem Netzwerk (Bandbreite, Latenz etc.).
Die Hersteller müssen die Basissicherheit und wesentlichen Leistungsmerkmale (siehe IEC 60601-1) gewährleisten.
Zudem betont die FDA, dass die Hersteller die Risiken über den kompletten Produktlebenszyklus (also nicht nur bei der Entwicklung, sondern auch in der Post-Market-Phase) analysieren und beherrschen müssen.
D) Verifizierung und Validierung
Weiter nennt die FDA Beispiele dafür, was die Hersteller beim „Testen“ (der Verifizierung und Validierung) berücksichtigten sollten:
- Erkennung korrupter Daten
- Sicherer Betrieb auch bei Daten außerhalb von Wertebereichen
- Übergang in den sicheren Zustand
- Konformität mit Interoperabilitätsstandards (wenn diese festgelegt wurden)
- Autorisierung und Authentisierung
- Gebrauchstauglichkeit
- Auswirkungen auf das Netzwerk und andere Geräte und Systeme
- Diese Tests sollen die Hersteller in simulierten Nutzungsumgebungen durchführen.
E) „Labeling“
Beschriftungen, Gebrauchs- und Installationsanweisungen und weitere Elemente des „Labelings“ sollen dafür ebenfalls zur Minimierung von Risiken beitragen. Was das Labeling genau enthalten soll, beschreibt das Guidance Document im Kapitel VI Teil D.
F) „Consensus Standards“
Die FDA weist (empfehlend) auf die „Consensus Standards“ hin, ohne im Dokument direkt welche zu benennen. Vielmehr verlinkt sie die Liste dieser Standards. Beispiele für diese Standards finden Sie weiter unten gelistet.
Nutzen Sie dieses Kapitel V dazu, um Ihren Entwicklungsplan zu ergänzen bzw. auf Vollständigkeit zu prüfen!
Kapitel VI: ‚Recommendations for Contents of Pre-market Submissions‘
Im Kapitel VI beschreibt die FDA, welche Informationen sie bei der Einreichung für ‚interoperable medical devices‘ erwartet. Diese Informationen entsprechen im Wesentlichen den im Kapitel V genannten. Daher seien hier nur einige Besonderheiten dieses Kapitels erwähnt:
- Die Dokumentation sollte ggf. beinhalten: Zweck und Technologien der Schnittstelle, verwendete Standards, Art der ausgetauschten Daten, funktionale und nicht funktionale Anforderungen, ggf. Beschreibung der API
- Die ISO 14971 und die Forderungen der 21 CFR part 820.30(g) sind zu beachten.
- Bei kritischen Anwendungen wie Infusionspumpen kann es sein, dass alle Testspezifikationen und -ergebnisse einzureichen sind. Bei unkritischen genügen ggf. Zusammenfassungen.
- Das Labeling muss auch den Anforderungen des 21 CFR part 801 und 809 genügen.
- Das Labeling (gemeint ist wahrscheinlich v.a. die Gebrauchsanweisung) sollte die im Kapitel V in den Unterkapiteln A und B genannten Aspekte beschreiben. Auch sollten den Anwendern Möglichkeiten gegeben werden, das korrekte Funktionieren der Schnittstellen zu prüfen.
Die FDA hat Ihr QM-System weitgehend auf die ISO 13485 umgestellt. Das betrifft auch die Anforderungen des 21 CFR part 820.30. Lesen Sie dazu mehr hier.
3. Die Anforderungen erfüllen
a. Dokumente ergänzen
Das FDA Guidance Document ‚Interoperable Medical Devices‘ stellt Anforderungen an die Entwicklung und Dokumentation von Medizinprodukten. Sie können diese Anforderungen umsetzen, indem Sie die folgenden Schritte gehen:
- Zweckbestimmung ergänzen
Stellen Sie sicher, dass die Zweckbestimmung Ihrer Medizinprodukte die in diesem Guidance-Dokument genannten Punkte festlegt wie z.B.
– Klinischer Kontext
– Einzubindende Geräte
– Vorgesehene Anwender
– Zweck des Datenaustauschs - System-/Software Requirements Specification ergänzen
Ergänzen Sie die Anforderungsdokumente. Spezifizieren Sie darin die Systeme / Software als Blackbox. Nutzen Sie das Blackbox-Modell.
Lesen Sie mehr zu diesem Thema in den Artikeln über das Blackbox-Modell und Anforderungen
Nutzen Sie Consensus Standards wo immer möglich. Mehr dazu lesen Sie weiter unten. - Gebrauchs- und Installationsanweisungen ergänzen
Mit diesen Festlegungen ergänzen Sie Ihre Anweisungen für die Anwender. Nutzen Sie Kapitel VI D als Checkliste, um die Vollständigkeit zu gewährleisten. - Risikomanagementakte ergänzen
Ergänzen Sie nun die Risikomanagementakte. Das betrifft die Risikotabelle ebenso wie den Risikomanagementplan, der auch die nachgelagerte Phase beinhalten muss. Ggf. passen Sie den Post-Market Surveillance Plan an. Er soll dazu verpflichten, gezielt und proaktiv nach Informationen über Risiken durch Interoperabilitätsprobleme zu suchen. - Produkt testen
Führen Sie die Verifizierungs- und Validierungsaktivitäten durch. Die System bzw. Software Requirements Specification und das Kapitel VI C dienen Ihnen dabei als Input.
Eine große Herausforderung wird darin bestehen, Systemumgebungen zu schaffen, die repräsentativ für die echten Umgebungen sind.
b. „Consensus Standards“ einhalten
Die FDA hat eine umfangreiche Liste an Consenus Standards publiziert wie die folgenden:
- ISO/IEEE 11073–10417 Third edition 2017–04 Health informatics—Personal health device communication—Part 10417: Device specialization—Glucose meter.
- ISO/IEEE 11073–10418 First edition 2014–03–01 Health informatics—Personal health device communication—Part 10418: Device specialization: International Normalized Ratio (INR) monitor [including TECHNICAL CORRIGENDUM 1 (2016)].
Diese Liste wurde, wie Sie am Datum erkennen, auch kürzlich aktualisiert.
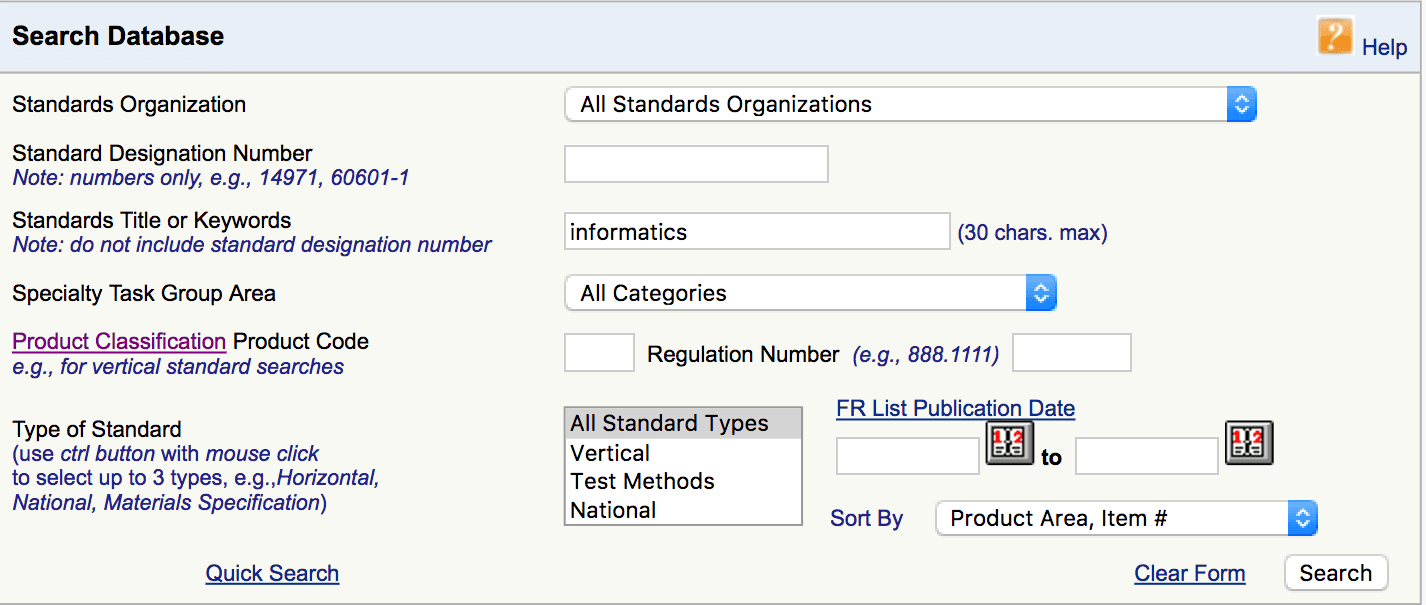
Sie finden diese Consenus Standards auf der Seite der FDA, in dem Sie dort im Feld „Standards Title or Keywords“ das Schlagwort „Informatics“ eingeben:
Überlassen Sie die Sicherheit Ihrer Patienten nicht dem Zufall
Gehen Sie mit einem Pentest des Johner Instituts auf Nummer sicher!
4. Zusammenfassung und Fazit
Das FDA Guidance Document zu den „Interoperable Medical Devices“ ist in die Jahre gekommen. Dennoch ist es weiterhin nützlich. Aktuellere Gedanken der FDA zur Interoperabilität finden Sie auf der FDA-Webseite „Medical Device Interoperability“.
Dass die FDA nur von „legally non binding“ und von „recommendation“ spricht, ist irreführend. Schließlich erwartet die US-Behörde bei Einreichungen genau diese in diesem Dokument genannten Informationen.
Das Guidance Document nennt keine „Consensus Standards“, sondern verweist auf die Liste der publizierten. Das macht das Dokument „wartbar“. Die Liste der Consensus Standards ist aber (zu) sehr auf (physische) Medizingeräte und die Normenfamilie ISO/IEC 11071 beschränkt. Für Stand-alone-Software hätte man sich mehr Hilfestellung gewünscht.
Änderungshistorie
- 2025-03-09: Dokument aktualisiert, Kapitelstruktur geändert
- 2017-09-26: Erste Version des Dokuments veröffentlicht


