
Die Cybersecurity von Medizinprodukten ist nicht nur ein Schwerpunkt der FDA, sondern auch anderer Gesetzgeber und Behörden, sowohl in den US als auch anderen Märkten.
Das ist nachvollziehbar,
- weil einerseits die Cybersecurity-Bedrohungen ständig zunehmen und
- andererseits die Medizinprodukte immer vernetzter und damit angreifbarer sind.
- Zudem ist die Patientenversorgung wie die Diagnose und Therapie immer stärker von diesen vernetzten Produkten abhängig.
Die USA hat den „Food, Drug & Cosmetic Act“ (FD&C) um Anforderungen an „Cyber Devices“ ergänzt, und die FDA mehrere Guidance Documents zur Cybersecurity veröffentlicht, die dieser Artikel vorstellt.
1. FDA Guidance zur Pre-Market Cybersecurity
Das erste Dokument umfasst 57 Seiten und trägt den Titel „Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submission“. Es wurde im September 2023 freigegeben. Allerdings hat die FDA bereits im März 2024 ein Update dazu geplant (siehe Kapitel 1.4 „Geplante Updates“).
Sie können sich die FDA Guidance zur Pre-Market Cybersecurity hier herunterladen.
1.1 Anforderungen an den Produktlebenszyklus
Eine zentrale Anforderung der FDA besteht darin, dass der Hersteller die Cybersecurity im QM-System verankert haben muss. Sie beschreit dazu einen Security Product Development Framework SPDF.
Die FDA nennt als weitere nützliche Quellen für das Verständnis des SPDF:
- den Joint Security Plan (Version 2 erschienen 03/2024)
- die IEC 81001-5-1
1.1.1 Ziele des SPDF
Dieses SPDF muss den ganzen Produktlebenszyklus abbilden und die „üblichen“ Ziele der IT-Sicherheit erreichen, Vertraulichkeit (Confidentiality), Integrität (Integrity) und Verfügbarkeit (Availability). Die FDA ergänzt diese „CIA-Ziele“ um die Fähigkeit des Produkts und Systems zum raschen Update bzw. Patch.
1.1.2 Vom SPDF geforderte Aktivitäten
Die FDA möchte, dass die Hersteller die Cybersecurity ihrer Produkte erreichen, indem sie ein „Security Risk Management“ durchführt. Dazu verweist sie auf die die AAMI TIR 57.
Ein Artikel zum AAMI TIR 57 beschreibt den Security Risk Management Prozess und dessen Zusammenspiel mit dem Risikomanagementprozess nach ISO 14971.
Als Teil dieses Prozesses müssen die Hersteller die folgenden Aktivitäten ausführen:
- Ein Threat Modelling des ganzen Systems, was eine System-Architektur voraussetzt
- Eine Bewertung der Cybersecurity-Risiken
- Die Bewertung der Risiken durch die Interoperabilität (dazu verweist die FDA auf das Guidance Document zur Interoperabilität)
- Eine Bewertung der Risiken durch Off-the-shelf-Software und das Erstellen einer Software Bill of Materials SBOM (mehr dazu im Fachartikel zur SBOM)
- Die Bewertung der Security-Risiken durch verbliebene „Anomalien“
- Eine fortlaufende Bewertung der Cybersecurity-Risiken in der Post-Market-Phase. Diese können sich ändern, weil beispielsweise neue Angriffsvektoren bekannt werden oder Produkte nicht mehr gepflegt werden.
- Auch die Überwachung des „Known Exploited Vulnerability Catalogs“ zählt dazu.
- Ein Cybersecurity Testing, das die Wirksamkeit von Maßnahmen zur Minimierung der Cybersecurity-Risiken prüft und beispielsweise ein Penetration- und Fuzz-Testing, eine statische und dynamische Code-Analyse sowie eine „Attack Surface Analysis“ beinhaltet.
1.2 Anforderungen an das Produkt
Im Anhang nennt die FDA auch konkrete Produktanforderungen mit Bezug zur Cybersecurity. Diese betreffen die Kryptografie, Authentifizierungs- und Autorisierungskonzepte, die Fähigkeit Angriffe zu protokollieren sowie das Produkt zu patchen.
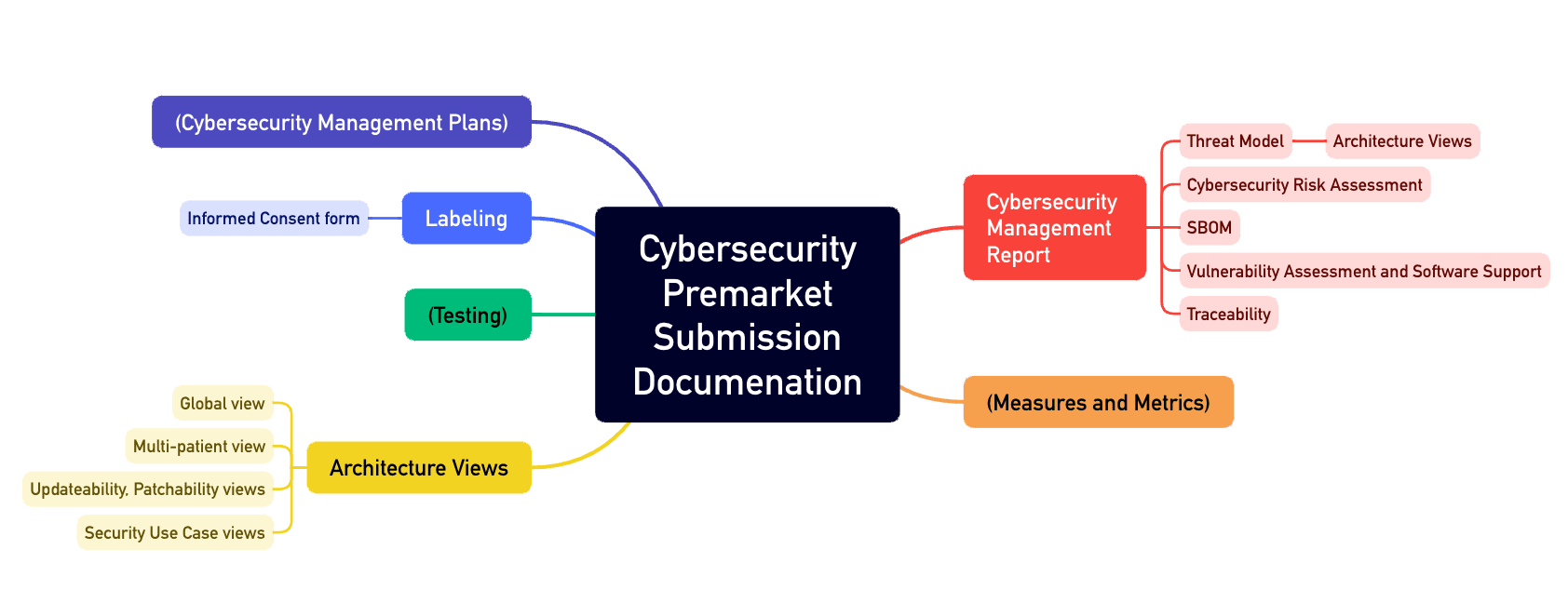
1.3 Anforderungen an die Dokumentation
All diese Tätigkeiten müssen in einer umfangreichen Dokumentation münden, wie beispielsweise:
- Cybersecurity Management Plan
- Architekturdokumentation mit verschiedenen „Views“ wie dem „Multi-Patient Harm View“ und dem „Updateability and Patchability View“
- SBOM
- Begleitmaterialien und Gebrauchsanweisungen
Selbst die Veröffentlichung von „Security Issues“ regelt die FDA und verweist dazu auf eine Vorgabe der NEMA mit dem Titel „Manufacturer Disclosure Statement for Medical Device Security“.
Der Anhang 4 des Guidance Documents beschreibt den Aufbau einer solchen Cybersecurity-Akte (s. Abb. 1).
1.4 Geplantes Update
Nach nur gut einem halben Jahr sieht die FDA die Notwendigkeit, das Dokument zu ergänzen. Diese geplanten Änderungen hat sie in einem Entwurf publiziert. Ein Grund ist der geänderte FD&C mit dem Abschnitt 524b.
1.4.1 Änderung bei der Definition
Die FDA gibt nun genauer vor, was sie als Cyber Device zählt: Bereits ein USB-Anschluss oder eine serielle Schnittstelle würden die Fähigkeit des Geräts begründen, sich mit dem Internet zu verbinden. Folglich müssten die Anforderungen der Abschnitts 524B des FD&C greifen und damit die Anforderungen der FDA an die Cybersecurity von Medizinprodukten.
1.4.2 Erweiterter Cybersecurity Management Plan
Zudem wünscht die FDA einen noch granulareren Cybersecurity Management Plan:
- Er soll eine genau Beschreibung enthalten, wie der Hersteller Patches entwickelt, freigibt und ausrollt.
- Ebenso regelt sie noch feiner, wie die Hersteller Schwachstellen und „Exploits“ offenbaren müssen.
- Schließlich soll der Plan, der über den kompletten Lebenszyklus gepflegt werden muss, unterscheiden, ob Produkte weiter gepflegt werden oder abgekündigt wurden, aber noch genutzt werden.
1.4.3 Änderungen bei Änderungsmitteilungen
Auch die Änderungsmitteilungen an die FDA sind von der geplanten Erweiterung des Guidance Documents zur Pre-Market-Cybersecurity betroffen. So will die Behörde sehen, welche Teile des Produkts geändert werden, die eine Auswirkung auf die Cybersecurity haben können oder sollen. Beispiele sind die Architektur, Verschlüsselungsalgorithmen und neue „Connectivity Features“.
1.4.4 Sonstige Änderungen
Relevant ist für die FDA künftig auch die Cybersecurity bei der Entscheidung, ob ein Produkt „substantially equivalent“ ist. D.h. Hersteller müssen explizit die Cybersecurity betrachten, wenn sie ein „predicate device“ angeben.
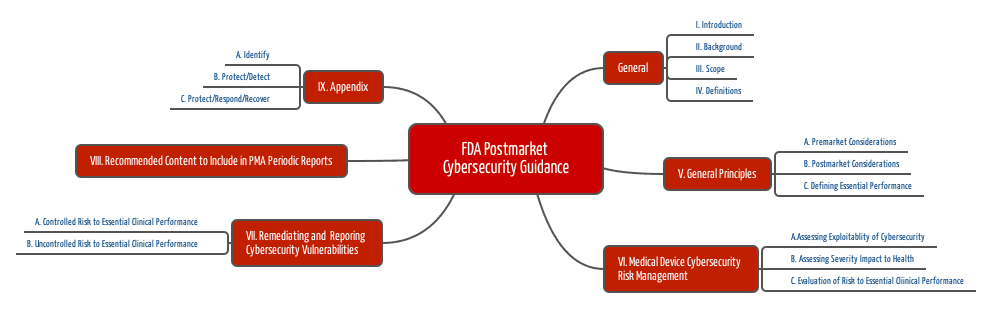
2. Post-Market Management of Cybersecurity
Deutlich in die Jahre gekommen ist eine zweite Guideline mit dem Titel „Post-Market Management of Cybersecurity in Medical Devices“ aus dem Jahr 2016.
Sie können diese „Post-Market Cybersecurity Guidance“ hier herunterladen.
Die FDA hat erkannt, dass Hersteller während der Entwicklung nicht alle künftigen Bedrohungen mit Bezug zur Cybersecurity vorhersagen und adressieren können. Daher verlangt sie, dass die Hersteller auch nach der Entwicklung kontinuierlich diese Bedrohungen analysieren und entsprechende Maßnahmen ergreifen.
Die Anforderungen des Pre-Market Guidance Dokuments enthält auch Anforderungen an die Post-Market Cybersecurity.
2.1 „Postmarket“-Informationsquellen
Zu den Informationsquellen, die die Hersteller auswerten sollen, zählen:
- Ergebnisse der Sicherheitsforscher
- Eigene Tests
- Soft- und Hardware-Lieferanten
- Kunden wie Krankenhäuser
- Institutionen, die sich auf das Sammeln, Analysieren und Verbreiten entsprechender Informationen spezialisiert haben
2.2 Maßnahmen
Die FDA empfiehlt das NIST Framework (National Institut for Standards and Technology) anzuwenden, dessen Elemente (Identify, Protect, Detect, Respond und Recover) auch das erste Guidance-Dokument aufgreift.
- Finden Sie heraus, welche wesentlichen (klinischen) Leistungsmerkmale Ihr Medizinprodukt erfüllen muss.
- Identifizieren Sie die Risiken, die auftreten, wenn diese Leistungsmerkmale nicht erfüllt sind — hier aufgrund eines Cybersecurity-Problems.
- Analysieren Sie, wie es zu diesem Problem kommen kann. Nutzen Sie dazu die o. g. Informationen.
- Bewerten Sie diese potenziellen Probleme und deren Wahrscheinlichkeiten, z. B. mit dem Common Vulnerability Scoring System.
- Analysieren Sie die Auswirkungen auf die Gesundheit, d. h. den Schweregrad möglicher Schäden.
- Bewerten Sie die Vertretbarkeit der Risiken.
- Beseitigen Sie Schwachstellen (immer), stellen Sie die Wirksamkeit der Maßnahmen sicher und dokumentieren Sie all das.
- Informieren Sie die Nutzer.
An dieser Stelle erwähnt die FDA, dass proaktive Maßnahmen zur Verbesserung der Cybersecurity gemäß 21 CFR part 806.10 der FDA nicht gemeldet werden müssen.
2.3 Zwischenfazit
Die Forderungen der FDA konkretisieren, was man von einer üblichen Marktüberwachung erwarten würde. Bemerkenswert sind:
- Das Dokument ist an einigen Stellen erstaunlich konkret, z. B. die auszuwertenden Informationsquellen bzw. die anzuwendenden Methoden betreffend.
- Das Dokument gibt Antworten auf die Fragen, wann Cybersecurity-bezogene Maßnahmen zu melden sind.
- Das Dokument definiert Begriffe (z. B. Exploit, Remediation, Threat, Threat Modelling) und gibt zahlreiche Beispiele.
3. Wesentliche Unterschiede zu Europa
3.1 Lebenszyklusphasen
Die Post-Market-Anforderungen will die FDA in einem Plan des Herstellers abgebildet sehen („Cyber Management Plan“). Diese Anforderungen stellt sie interessanterweise im Pre-Market Guidance Document.
In Europa erwartet man hingegen bei Harmonisierung der IEC 81001-5-1, dass das QM-System alle Pre- und Postmarketprozesse vollständig abbildet.
3.2 Dokumentation
Die FDA ist bei der Forderung nach einer SBOM wesentlich präziser: Während die IEC 81001-5-1 und das MDCG 2019-16 die SBOM bestenfalls erwähnen, verlinkt die FDA auf die NTIA-Spezifikation und ergänzt noch weitere Forderungen (z.B. Angabe des End of Support zu jedem Element der SBOM).
Die FDA legt großen Wert auf die „architectural views“ und spezifiziert sehr präzise ihre Erwartungen.
3.3 Sonstiges
Zudem erwartet die FDA Metriken, um die Reaktionsfähigkeit des Herstellers auf bekanntgewordene Schwachstellen zu bewerten.
Mit der konkreten Liste an Artefakten zur Einreichung und der Aussage, dass der Begriff „cyber device“ sich möglicherweise auf alle Datenschnittstellen bezieht (explizit auch auf USB), ist die FDA ebenfalls sehr deutlich mit ihren Erwartungen. Die EU lässt mit MDCG 2019-16 und IEC 81001-5-1 deutlich mehr Spielraum bei der Interpretation.
4. Tipps für Hersteller
Die Erfahrungen des Johner Instituts zeigen, dass Hersteller von den folgenden Tipps profitieren:
- Nutzen Sie das eStar-Format. Denn es listet die konkreten Artefakte der Cybersecurity, die die FDA bei der Einreichung bei allen Zulassungsverfahren (auch PMA) erwartet.
- Beachten Sie, dass selbst rein lokale Schnittstellen (z.B. USB oder Bluetooth) eine vollständige Cybersecurity-Dokumentation erfordern. Allerdings ist eine Skalierung des Umfangs dieser Dokumentation möglich.
- Nutzen Sie bei Unklarheiten „Pre-Submission-Meetings“ mit der FDA. Die Behörde ist oft überraschend hilfsbereit und offen für Diskussionen.
- Greifen Sie auf unsere Hilfe zurück, insbesondere wenn Sie noch wenig Erfahrungen mit dem Thema gesammelt haben. Denn der Weg des Eigenstudiums ist meist aufwendig und fehlerbehaftet.
- Lassen Sie Penetration-Test extern durchführen (z.B. bei uns), um keine Diskussionen um die Unabhängigkeit der Tester zu riskieren. Lassen Sie die zumindest die finale Software-Konfiguration testen. Da wir dabei fast immer Schwachstellen finden, empfehlen wir entwicklungsbegleitende kleinere Pen-Tests.
- Das Threat-Modeling ist ebenfalls sehr aufwendig, zeitintensiv und bedarf großer Erfahrung mit Security-Risiken. Deshalb empfehlen wir auch hier Tools zu nutzen und Experten einzubinden.
5. Fazit und Zusammenfassung
Medizinprodukte, die Software enthalten oder Software sind, verfügen meist über Datenschnittstellen. Deshalb müssen sie ausreichend cybersicher ausgelegt werden.
Die FDA beschreibt ihre Anforderungen recht detailliert in ihren beiden Guidance-Dokumenten für Pre- und Postmarket. Darüber hinaus gibt sie mit den Recognized Consensus Standards praktische Hilfen zur Umsetzung.
Erfreulich ist, dass diese Liste auch die IEC 81001-5-1 erwähnt, weil diese Norm in anderen Teilen der Welt bereits verbindlich (z.B. Japan) oder harmonisiert (in der EU in Arbeit) ist.
Mit der neusten geplanten Anpassung der Premarket Guidance legt die FDA die Messlatte noch höher. Denn jetzt kommen auch bei Änderungen an bereits zugelassenen Produkten Anforderungen aus der Cybersecurity auf die Hersteller zu.
Nutzen Sie die Hilfe des Johner Instituts bei der Interpretation und effizienten Umsetzung von FDA-Anforderungen. Nehmen Sie gleich Kontakt auf mit den IT-Security-Experts des Johner Instituts
Änderungshistorie
- 2024-03-26: Artikel komplett neu geschrieben mit Ausnahme des zweiten Kapitels
- 2018-10-23: Erste Version des Artikels publiziert
Überlassen Sie die Sicherheit Ihrer Patienten nicht dem Zufall
Gehen Sie mit einem Pentest des Johner Instituts auf Nummer sicher!


Sehr Hilfreich!
Hallo Herr Rosenzweig,
zunächst herzlichen Dank für Ihre Mühe, die Informationen der FDA-Guidances so übersichtlich und hilfreich darzustellen! Kann es sein, dass der Draft der Guidance von 2018 von der FDA zurückgezogen wurde? Es gibt wohl seit April 2022 einen neuen Draft (aber mit etwas verändertem Titel), der die bisher noch gültige Guidance von 2014 ablösen soll. Mich würde interessieren, ob der Inhalt, den Ihr Artikel zum Draft von 2018 beschreibt (Thema CBOM, Tiers etc.) dennoch aktuell ist?
Vielen Dank und Grüße,
Beatrice Dachsel
Liebe Frau Dachsel, tatsächlich hat der neuste Guidance-Entwurf von April 2022 denjenigen von 2018 schon wieder obsolet gemacht. Einige wichtige Erkenntnisse der FDA sind in den neusten Entwurf eingeflossen. So hat man sich von der Abkürzung CBOM verabschiedet und die international gängige Abkürzung SBOM eingeführt. Damit einhergehend liegt der Schwerpunkt jetzt auf den Software-Elementen, die im Medizinprodukt verwendet werden. Auch das Tier-Konzept, das in der letzten Version noch aufgeführt war, ist verschwunden. Wir werden unseren Artikel also nochmals überarbeiten und weitere Gesichtspunkte dabei aufführen. Geben Sie uns dafür bitte noch etwas Zeit.
Herzliche Grüße
Christian Rosenzweig
Vielen Dank für Ihre schnelle Antwort!