Entspricht ein Medizinprodukt nicht (mehr) den Vorgaben der FDA, müssen sich Hersteller, Händler und Importeure einen Rückruf (Recall) durchführen. Das gilt insbesondere, wenn von dem Produkt eine Gefahr ausgeht.
In diesem Beitrag erfahren Sie,
- was ein Recall (Rückruf) ist,
- welche Varianten es gibt,
- was es dabei zu beachten gibt,
- was der Unterschied zwischen „Recall“ sowie „Corrections and Removal“ ist und
- wer den Recall bzw. die Correction and Removal durchführen muss.
1. Was ein Recall ist
1.1 Definition der FDA des Begriffs Recall
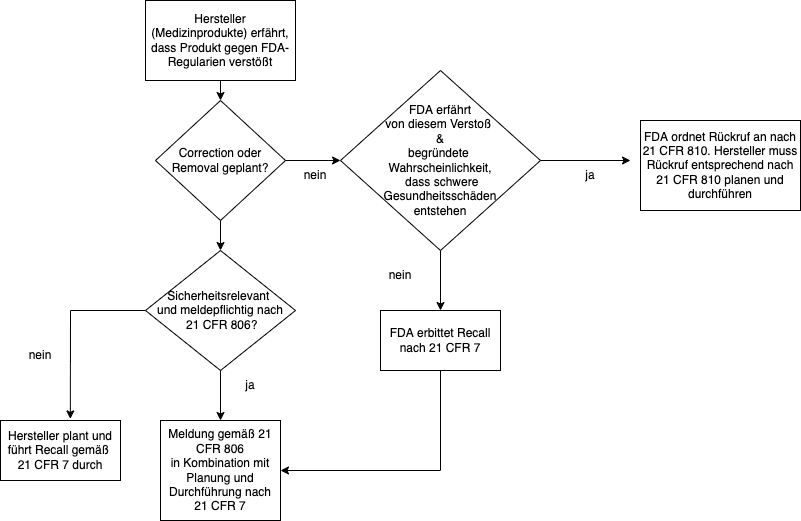
Mit einem Rückruf (Recall) korrigieren Hersteller ihre Produkte oder nehmen diese vom Markt, wenn sie nicht (mehr) den Vorgaben der FDA entsprechen. Hersteller und Händler kommen damit ihrer Verantwortung nach, die Gesundheit und das Wohlergehen der Öffentlichkeit vor Produkten zu schützen, die eine Verletzungsgefahr darstellen, grobe Täuschung beinhalten oder anderweitig fehlerhaft sind.
Rücknahme oder Korrektur eines vermarkteten Produkts durch ein Unternehmen, das nach Ansicht der FDA gegen die von ihr verwalteten Gesetze verstößt und gegen das die Behörde rechtliche Schritte einleiten würde, z. B. die Beschlagnahme. Ein Rückruf umfasst nicht die Rücknahme vom Markt oder die Rückgewinnung von Lagerbeständen.
Quelle: FDA
1.2 Recall-Varianten
Die FDA unterscheidet drei verschiedene Arten von Recalls:
- Rückrufe von Medizinprodukten werden in der Regel vom Hersteller gemäß 21 CFR 7 freiwillig durchgeführt.
- Ein Rückruf kann auch von der FDA angeordnet werden. Dies geschieht, wenn der Hersteller oder Importeur ein gesundheitsgefährdendes Produkt nicht freiwillig vom Markt nimmt. Die Rückrufaufforderung richtet sich nach 21 CFR 810 und wird von der Medical Device Recall Authority erteilt.
- Eng mit dem Rückruf verwandt ist die Correction and Removal (Korrektur und Entfernung) nach 21 CFR 806. Diese erfolgt ebenfalls auf Veranlassung des Herstellers selbst, etwa um sicherzustellen, dass das Produkt weiterhin gesetzeskonform ist. Eine Correction-and-Removal-Maßnahme ist jedoch unter bestimmten Umständen meldepflichtig.
Eine detaillierte Übersicht über Rückrufmaßnahmen gibt es auf den Seiten der FDA.
2. Freiwilliger Recall nach 21 CFR 7 (Enforcement Policy)
Der Rückruf von Medizinprodukten nach 21 CFR 7 erfolgt freiwillig durch den Hersteller oder den Händler. Dieser ruft ein auf dem Markt befindliches Produkt dann zurück, wenn es gegen rechtliche Vorgaben verstößt. Dies gilt insbesondere für Produkte, die die Gesundheit und das Wohlergehen der Öffentlichkeit gefährden, weil
- sie eine Verletzungsgefahr darstellen,
- sie grobe Täuschung beinhalten
- oder anderweitig fehlerhaft sind.
Beachten Sie das Guidance-Dokument der FDA über Volontary Recalls nach 21 CFR Part 7, Subpart C.
2.1 Was der freiwillige Rückruf NICHT umfasst
Ein Rückruf nach 21 CFR 7 umfasst nicht die Rücknahme vom Markt oder die Rücknahme von Lagerbeständen.
‚Marktrücknahme‘ bedeutet, dass ein Unternehmen ein vertriebenes Produkt zurücknimmt oder korrigiert, wenn es sich um einen geringfügigen Verstoß handelt, der von der FDA nicht geahndet wird. Hierunter fallen auch Rückrufe, die ohne Verstoß erfolgen, z. B. normale Lagerumschlagspraktiken, routinemäßige Anpassungen oder Reparaturen von Geräten.
Quelle: FDA
Stellt die FDA jedoch fest, dass das zurückgenommene Produkt doch gegen Vorschriften verstößt, gelten die Regeln für Rückrufe. Ist ein Unternehmen unsicher, ob ein Produkt gegen FDA-Vorschriften verstößt, muss es die Rücknahme vom Markt der FDA melden.
Außerdem unterliegen elektronische Produkte, die Strahlung aussenden und unter 21 CFR 1003 und 1004 fallen, nicht den Anforderungen von 21 CFR 7.
2.2 Requested Recall nach 21 CFR 7
Kümmern sich weder Hersteller noch Händler freiwillig um einen Rückruf ihres Produkts, das den Vorgaben nicht entspricht, und erfährt die FDA davon, kann sie einen Rückruf auch nach 21 CFR 7 anfordern (‚requested recall‘). Anders als beim angeordneten Rückruf von Medizinprodukten nach 21 CFR 810 ist dies bei 21 CFR 7 in Ausnahmefällen und bei hoher Dringlichkeit vorgesehen.
Die Aufforderung richtet sich in diesem Fall an den Hersteller. Die Maßnahmen der FDA reichen von einer reinen Aufforderung bis hin zur Beschlagnahmung in Extremfällen.
A request by the Food and Drug Administration that a firm recalls a product is reserved for urgent situations and is to be directed to the firm that has primary responsibility for the manufacture and marketing of the product that is to be recalled.
2.3 Vorgehen beim freiwilligen Rückruf
2.3.1 Schritt 1: Meldung des Rückrufs an die FDA
Ein Rückruf nach 21 CFR 7 muss an die FDA gemeldet werden. Hierfür müssen sich Hersteller an den Division Recall Coordinator (DRC) ihres FDA Office of Regulatory Affairs (ORA) wenden. Hersteller aus dem Ausland und Importeure müssen sich an das DRC wenden, in dem ihr US-Agent ansässig ist.
Der Rückruf soll so schnell wie möglich erfolgen. Eine Frist ist nicht vorgegeben.
2.3.2 Schritt 2: Bewertung der Gesundheitsgefährdung durch die FDA
Die FDA klassifiziert Rückrufe mit einer numerischen Bezeichnung (I, II oder III), um den relativen Grad der Gesundheitsgefährdung durch das zurückgerufene Produkt anzugeben.
- Klasse I – Eine Situation, in der eine begründete Wahrscheinlichkeit besteht, dass die Verwendung eines nicht-konformen Produkts oder die Exposition gegenüber einem solchen Produkt schwerwiegende gesundheitsschädliche Folgen oder den Tod zur Folge hat.
- Klasse II – Eine Situation, in der die Verwendung eines verletzenden Produkts oder die Exposition gegenüber einem verletzenden Produkt vorübergehende oder medizinisch reversible gesundheitsschädliche Folgen haben kann oder in der die Wahrscheinlichkeit ernsthafter gesundheitsschädlicher Folgen gering ist.
- Klasse III – Eine Situation, in der die Verwendung eines gesundheitsschädlichen Produkts oder die Exposition gegenüber einem solchen Produkt wahrscheinlich keine gesundheitsschädlichen Folgen hat.
Je nach Klasse ist der interne Prozess der FDA unterschiedlich. Sämtliche Recalls der Klasse I werden auf der FDA-Website gepostet. Somit sollte die Klasse auch einen Einfluss auf die Recall-Strategie haben.
Bei hohem Risiko (Klasse I) sollten Hersteller im Rahmen der Strategie strengere Maßnahmen festlegen, z. B. 100-prozentige Wirksamkeit des Rückrufs, 100 Prozent Antwortrate der Kunden, mehr Versuche, sämtliche Kunden zu kontaktieren, Meldung über verschiedene Medien etc.
2.3.3 Schritt 3: Rückrufstrategie festlegen
Das rückrufende Unternehmen muss eine Rückrufstrategie entwickeln, die die FDA genehmigen muss. Diese muss den möglichen Folgen der möglichen Gefährdung durch das Produkt entsprechen.
‚Rückrufstrategie‘ bezeichnet eine geplante Vorgehensweise bei der Durchführung eines bestimmten Rückrufs, die sich auf die Reichweite des Rückrufs, die Notwendigkeit öffentlicher Warnungen und den Umfang der Wirksamkeitskontrollen für den Rückruf bezieht.
Quelle: FDA
Die FDA nennt für die Ausgestaltung der Strategie die folgenden Kriterien:
- Ergebnisse der Bewertung der Gesundheitsgefährdung (die Klasse hat Einfluss auf die Strategie; z. B. müssen Unternehmen bei hoher Gefährdung schneller und entschiedener handeln)
- Leichte Identifizierbarkeit des Produkts
- Grad der Offensichtlichkeit des Produktmangels für den Verbraucher oder Anwender
- Ausmaß, in dem das Produkt auf dem Markt unbenutzt bleibt
- Kontinuierliche Verfügbarkeit von wichtigen Produkten
Die Rückrufstrategie muss (je nach dem zuvor festgestellten Schweregrad des Verstoßes) die folgenden Elemente berücksichtigen:
- Reichweite des Rückrufs
Hier sind die Vertriebsketten zu berücksichtigen, auf die sich der Rückruf erstrecken soll: Verbraucher- oder Benutzerebene oder Groß- bis Einzelhandelsstufe. Wie weit der Rückruf geht, hängt vom Gefährdungsgrad durch das Medizinprodukt ab. - Öffentliche Warnung – ja oder nein?
Besteht eine besonders ernste Gefahr, kann auch eine öffentliche Warnung nötig sein. Diese ist mit der FDA abzustimmen und wird in der Regel von dieser selbst vorgenommen. Eine öffentliche Warnung ist jedoch dringenden Situationen vorbehalten. - Wirksamkeitskontrolle
Das rückrufende Unternehmen muss Strategien zur Wirksamkeit der Rückrufmaßnahmen etablieren. Laut FDA sind hierfür etwa persönliche Besuche, Telefonanrufe, Briefe oder eine Kombination davon angedacht.
‚Rückrufendes Unternehmen‘ ist das Unternehmen, das einen Rückruf veranlasst, oder – im Falle eines von der Food and Drug Administration angeordneten Rückrufs – das Unternehmen, das die Hauptverantwortung für die Herstellung und Vermarktung des zurückzurufenden Produkts trägt.
Quelle: FDA
2.3.4 Schritt 4: Recall Letter versenden
Ein Unternehmen, das einen Rückruf durchführt, muss jeden betroffenen Direktkunden unverzüglich über den Rückruf informieren. Format, Inhalt und Umfang einer Rückrufmitteilung richten sich nach der Gefahr, die vom Produkt ausgeht. Kunden sind ihrerseits verpflichtet, der Rückrufaufforderung unverzüglich nachzukommen.
Die FDA gibt Tipps für das Verfassen der Mitteilung:
- Fassen Sie sich kurz und seien Sie prägnant.
- Geben Sie das Produkt, die Größe, die Identifikationsnummer (z. B. UDI) und alle anderen sachdienlichen beschreibenden Informationen eindeutig an, um eine genaue und sofortige Identifizierung des Produkts zu ermöglichen.
- Erläutern Sie kurz und bündig den Grund für den Rückruf und die damit verbundene Gefahr, falls vorhanden.
- Geben Sie genaue Anweisungen, was mit den zurückgerufenen Produkten zu tun ist.
- Stellen Sie dem Empfänger eine Möglichkeit bereit, dem rückrufenden Unternehmen mitzuteilen, ob er das Produkt besitzt.
- Die Rückrufmitteilung sollte keine irrelevanten Informationen, Werbematerialien oder andere Aussagen enthalten, die von der Botschaft ablenken könnten.
2.3.5 Schritt 5: Über den Stand des Rückrufs berichten
Das rückrufende Unternehmen soll dem Division Recall Coordinator (DRC) regelmäßig Berichte über den Stand des Rückrufs übermitteln, damit die FDA den Fortschritt des Rückrufs beurteilen kann. Im Allgemeinen beträgt der Berichtszeitraum zwischen zwei und vier Wochen.
2.4 Veröffentlichung und Beendigung eines Rückrufs
2.4.1 Beendigung des Rückrufs
Ein Rückruf wird beendet, wenn die FDA feststellt, dass alle angemessenen Anstrengungen unternommen wurden, um das Produkt in Übereinstimmung mit der Rückrufstrategie zu entfernen oder zu korrigieren. Der DRC benachrichtigt das rückrufende Unternehmen über die Beendigung schriftlich. Das Unternehmen selbst kann die Beendigung des Rückrufs auch beantragen.
2.4.2 Öffentliche Bekanntgabe des Rückrufs
Die FDA veröffentlicht Rückrufe in ihrem wöchentlichen FDA-Enforcement-Bericht.
- Handbuch für Regulierungsverfahren, Chapter 7 Recall Procedures
- Medical Device Class I Recalls
- Rückrufe von Geräten: Eine Studie zu Qualitätsproblemen, Dokument Nr. 273, Kontakt: dice@cdrh.fda.gov „Evaluation of Software Related Recalls for Fiscal Years 1983-91“, Mai 1992, Kontakt: dice@cdrh.fda.gov
3. Nutzungsuntersagung nach 21 CFR 810 – Medical Device Recall Authority
Entsprechen Medizinprodukte nicht (mehr) den FDA-Vorgaben, werden sie in der Regel freiwillig gemäß 21 CFR 7 zurückgerufen. In Ausnahmefällen, in denen der Hersteller oder Importeur ein gesundheitsgefährdendes Produkt nicht freiwillig zurückruft, ergreift die FDA Zwangsmaßnahmen gemäß 21 CFR 810 – Medical Device Recall Authority.
Dies geschieht normalerweise,
- nachdem die FDA dem Unternehmen Gelegenheit gegeben hat, sich mit der Behörde zu beraten,
- das Unternehmen den Rückruf dennoch nicht freiwillig durchführt,
- die FDA zu dem Schluss kommt, dass eine begründete Wahrscheinlichkeit besteht, dass ein Produkt schwerwiegende gesundheitsschädliche Folgen oder den Tod verursachen würde.
In diesem Fall verbietet die FDA zunächst den Vertrieb und die Nutzung des Produkts und setzt davon auch mögliche Nutzer wie medizinische Einrichtungen in Kenntnis. Die Nutzungsuntersagung kann später auch in einen “Zwangsrückruf” gewandelt werden.
Liegen die Voraussetzungen von 21 CFR 810 vor, ist dieser vorrangig zu 21 CFR 7.
4. Korrektur und Rücknahme nach 21 CFR 806 – Medical Device Correction and Removals
4.1 Abgrenzung von Recall, Correction and Removal
Eng mit dem Rückruf (Recall) verknüpft ist die sogenannte Correction (Korrektur) nach 21 CFR 806. Der grundlegende Unterschied zwischen der Korrektur und dem Rückruf besteht darin, dass Korrekturen auch dann erfolgen, wenn das Produkt nicht gegen FDA-Vorschriften verstößt.
- Rückruf: Ein Rückruf ist eine Korrektur oder Entfernung, die erfolgt, wenn das Produkt gegen rechtliche Vorschriften verstößt.
- Korrektur: Das Produkt wird verbessert, angepasst, neu gekennzeichnet oder repariert. Dies muss nicht zwangsläufig erfolgen, um einen Verstoß gegen Regelungen zu korrigieren.
- Entfernung: Die physische Entfernung eines Produkts von seinem Verwendungsort zu einem anderen Ort zwecks Reparatur, Änderung, Einstellung, Neuetikettierung, Vernichtung oder Überprüfung.
Ein Rückruf kann also beides sein, Korrektur oder Entfernung. Er erfolgt jedoch immer aufgrund eines Verstoßes gegen rechtliche Vorschriften. Korrekturen und Entfernungen können auch unabhängig eines Verstoßes erfolgen.
on (einschließlich Patientenüberwachung) eines Produkts, ohne dass es physisch von seiner Verwendungsstelle an einen anderen Ort verbracht wird.
Quelle: 21 CFR 806.2 (d) und (j)) nach FDA
‚Entfernung‘ bedeutet die physische Entfernung eines Produkts von seiner Verwendungsstelle an einen anderen Ort zwecks Reparatur, Änderung, Einstellung, Neuetikettierung, Zerstörung oder Inspektion.
Quelle: 21 CFR 806.2 (d) und (j)) nach FDA
4.2 Wann die Korrektur meldepflichtig ist
Eine Korrektur ist normalerweise nicht meldepflichtig. Sie kann jedoch meldepflichtig werden, wenn sie erfolgt,
- um ein von dem Produkt ausgehendes Gesundheitsrisiko nach 21 CFR 806.2 (k) zu verringern. Die Definition des Gesundheitsrisikos entspricht den Definitionen für Rückrufe der Klassen I und II in 21 CFR 7.3(m) (siehe oben),
- um einen durch das Produkt verursachten Verstoß gegen den FD&C Act zu beheben, der ein Gesundheitsrisiko darstellen kann, es sei denn, die Informationen wurden bereits im Rahmen einer Anzeige von meldepflichtigen Vorkommnissen (Medical Device Reporting) gemäß 21 CFR 803 (MDR-Meldung) übermittelt oder die Korrektur- oder Beseitigungsmaßnahme ist von der Meldepflicht ausgenommen (21 CFR 806.1 (b).
- Eine Meldung muss auch dann erfolgen, wenn das Ereignis auf einen Anwendungsfehler zurückzuführen ist.
Hersteller und Importeure müssen Aufzeichnungen über die Korrekturen und Entfernungen führen, die nicht an die FDA gemeldet werden müssen. Wenn eine Meldung gemäß 21 CFR 806 nicht erforderlich ist, kann das Unternehmen eine freiwillige Meldung gemäß 21 CFR 7 vornehmen.
Ausnahmen von der Meldepflicht
Von der Meldepflicht in den o. g. Fällen sind die folgenden Maßnahmen ausgenommen (21 CFR 806.1 (b)):
- Maßnahmen zur Verbesserung der Leistung oder der Qualität eines Produkts
- Marktrücknahmen, die keinen oder nur einen geringfügigen Verstoß gegen das FDA-Gesetz darstellen, aber nicht Gegenstand rechtlicher Schritte der FDA wären, z. B. normale Lagerrotation
- Routinewartung (regelmäßig geplante Wartung eines Produkts einschließlich des Austauschs von Teilen am Ende ihrer normalen Lebensdauer, z. B. Kalibrierung, Austausch von Batterien und Reaktion auf normale Abnutzung)
- Rücknahme von Lagerbeständen
Sicherheitswarnungen/Feldsicherheitshinweise gelten nicht als meldepflichtige Korrektur und Entfernung, können aber Abhilfemaßnahmen sein.
4.3 Wie Correction and Removal erfolgen muss
4.3.1 Wer melden muss
Melden muss das Unternehmen, das die Korrektur veranlasst. Das können Hersteller oder Importeure sein.
4.3.2 Welche Fristen gelten
Der Bericht muss der FDA innerhalb von zehn Arbeitstagen ab dem Zeitpunkt vorliegen, an dem das Unternehmen die Korrektur oder Entfernung veranlasst hat.
Dieselbe Frist gilt auch für Änderungen der ursprünglichen Korrekturmeldung (zehn Arbeitstage ab Änderung).
4.3.3 Was in der Meldung stehen muss
Was Hersteller oder Importeure genau melden müssen, steht in § 806.10(c).
- Registrierungsnummer, Datum der Meldung, laufende Nummer (001, 002 usw.), „C“ für Correction (Korrektur) oder „R“ für Removal (Entfernung)
- Name, Adresse, Telefonnummer und Kontaktperson der Firma, die für die Durchführung der Korrektur oder Entfernung verantwortlich ist
- Markenname und gebräuchliche Bezeichnung des Produkts und Verwendungszweck
- FDA-Vermarktungsstatus, d. h. 510(k), PMA, Preamendment-Status und Nummer der Produktliste
- Modell-/Katalognummer, Los-/Seriennummer
- Kontaktinformationen des Herstellers (Name, Adresse, Telefonnummer, Kontaktperson), falls abweichend von Punkt 2 oben
- Beschreibung des Vorfalls/der Vorfälle und der Korrektur- und Beseitigungsmaßnahmen, die bereits ergriffen wurden bzw. noch ergriffen werden sollen
- Alle Erkrankungen oder Verletzungen, die im Zusammenhang mit der Verwendung des Produkts aufgetreten sind. Falls zutreffend, sind alle gemäß 21 CFR 803 eingereichten MDR-Nummern (Medical Device Report) anzugeben.
- Anzahl der Produkte, die von der Korrektur oder Entfernung betroffen sind
- Datum der Herstellung oder des Vertriebs, Verfallsdatum oder erwartete Lebensdauer
- Name, Anschrift und Telefonnummer aller Empfänger (im In- und Ausland) sowie Datum und Anzahl der an jeden Empfänger verteilten Produkte
- Eine Kopie aller Mitteilungen über die Berichtigung oder Entfernung
- Eine Erklärung, warum die erforderlichen Informationen nicht verfügbar sind, und ein Datum, an dem sie nachgereicht werden sollen
4.3.4 An wen sich die Meldung richtet
Hersteller und Importeure haben zwei Möglichkeiten, Korrekturen und Entfernungen zu melden:
- Über die FDA Electronic Submission Software (eSubmitter). Dies wird von der FDA empfohlen.
- Per E-Mail an den Division Recall Coordinator (DRC) der FDA Office of Regulatory Affairs (ORA).
5. Tipps & weiterführende Informationen
5.1 Tipps
Damit der Prozess des Rückrufs oder der Korrektur reibungslos abläuft, haben wir folgende Tipps für Hersteller und Importeure.
5.1.1 Sich der Verantwortung bewusst sein
Hersteller aus dem Ausland und Importeure sollten auf einen Recall bzw. eine Correction and Removal vorbereitet sein. Häufig unterliegen Unternehmen dem Irrglauben, dass ihr US-Agent die Meldung übernimmt. Doch Recalls, Corrections und Removals liegen in der Verantwortung des Herstellers bzw. des Importeurs.
5.1.2 Verfahren etablieren
Unternehmen, die ihre Produkte auf dem US-Markt vertreiben, sollten im Rahmen ihres QM-Systems ein Verfahren zum Thema Recall bzw. meldepflichtige Correction and Removal etablieren.
Der Prozessablauf sollte geübt werden, damit das Unternehmen im Ernstfall schnell reagieren kann.
5.1.3 Vorlagen für Meldungen erstellen
Hersteller und Importeure sollten Vorlagen erstellen, z. B. zur Meldung an die FDA, zur Meldung an die Anwender etc. Dies spart im Ernstfall Zeit und hilft, Fehler zu vermeiden.
Im Auditgarant finden Hersteller und Importeure Anleitungen und weitere wichtige Informationen zu Recalls und Corrections/Removals. Unter anderem gibt es eine Vorlage für ein Verfahren zu Recalls bzw. Corrections und Removals.
5.1.4 Klare Rollen und Vertretung etablieren
Legen Sie die Rollenverteilung für den Fall eines Recalls bzw. einer Correction and Removal im Vorfeld fest. Wichtig ist es auch, eine Vertreterregelung zu etablieren. Es darf nicht sein, dass ein meldepflichtiger Recall nicht bearbeitet werden kann, weil die Mitarbeitenden gerade in Urlaub sind.
5.1.5 21 CFR 806 beachten
Häufig denken Hersteller nur an den 21 CFR 803 (Medical Device Reporting), wenn es um die Meldung von Vorkommnissen geht. Ergreifen Hersteller in diesem Zusammenhang aber Korrekturmaßnahmen im Feld, ist unbedingt 21 CFR 806 (Medical Device Correction and Removals) zu befolgen. Versäumen Unternehmen dies, droht ein Warning Letter durch die FDA.
5.2 Guidances und weitere wichtige Literatur zu FDA-Recalls
Für weitere Recherche empfehlen wir die folgenden Ressourcen:
- Website der FDA zum Thema Recalls und Corretions/Removals
- FDA Guidance-Dokument Initiation of Voluntary Recalls Under 21 CFR Part 7, Subpart C (PDF)
- Guidance for Industry and FDA Staff (PDF)
- Auf der FDA-Plattform CDRH-Learn gibt es im Abschnitt Postmarket Activities / Medical Device Recalls zahlreiche Präsentationen und Webinare zu den Themen Recall sowie Correction and Removal.
- Regulatory Procedures Manual: Chapter 7 Recall Procedures (PDF)
6. Zusammenfassung und Fazit
Gerade Hersteller und Importeure sollten sich unbedingt mit dem Prozess für Recalls, Corrections und Removals auseinandersetzen. Gibt es in den USA Probleme mit ihrem Produkt, sind sie u. U. für das Verfahren verantwortlich. Bei Untätigkeit droht eine Abmahnung durch die FDA und schlimmstenfalls eine Klage.
Auf den ersten Blick wirkt der Prozess für Recalls sowie Corrections und Removals kompliziert und aufwändig. Doch wer sich gut vorbereitet und die genannten Hinweise befolgt, sollte im Ernstfall einen reibungslosen Ablauf erwarten können.
Wenn bei Ihnen ein Recall, eine Correction oder ein Removal im Raum steht, dann melden Sie sich gleich (z.B. über das Webformular). Unsere FDA Experts stellen sicher, dass Ihnen keine Fehler bei diesen gesetzlich sehr eng regelten Schritten unterlaufen.
Änderungshistorie
- 2024-02-25: Artikel neu strukturiert