
Die FDA beschreibt in zwei Dokumenten ihre Anforderungen an das Human Factors Engineering:
- Applying Human Factors and Usability Engineering to Medical Devices (Feb. 2016, finale Version)
- Content of Human Factors Information in Medical Device Submissions (Dez. 2022, Entwurf)
Dieser Artikel
- fasst für Sie die wichtigsten Anforderungen zusammen,
- enthält eine kommentierte Version des zweiten Dokuments zum Download und
- gibt Hilfestellungen, damit Sie diese Anforderungen schnell und sicher erfüllen und Probleme bei der Zulassung vermeiden können.
Was die FDA als „Human Factors Engineering“ (HFE) bezeichnet, nennt die IEC 62366-1 „Usability Engineering“.
Die FDA definiert den Begriff wie folgt:
Application of knowledge about human behavior, abilities, limitations, and other characteristics to the design of medical devices (including software), systems and tasks to achieve adequate usability.
Quelle: Content of Human Factors Information in Medical Device Submissions
1. Wie die „Human Factors Guidances“ der FDA zusammenspielen
In der Einleitung des Dokuments „Content of Human Factors Information in Medical Device Submissions“ beschreibt die FDA ihre Ziele:
- Dieses Dokument soll Herstellern eine Anleitung bieten, welche Informationen zu Human Factors (Usability) abhängig vom Risiko des Produkts bei der FDA bei einer Zulassung eingereicht werden müssen.
- Es soll die FDA-Guidance „List of Highest Priority Devices for Human Factors Review“ ersetzen.
- Es soll die FDA-Guidance „Applying Human Factors and Usability Engineering to Medical Devices“ (HFE-Guidance) ergänzen, präzisieren und die Kapitel 3 (Definitions), 9 (Documentation) und Annex A (HFE/UE Report) sogar ersetzen.
- Die HFE-Guidance definiert den HFE-Prozess, aus dem die Dokumente resultieren, die ganz oder teilweise bei der FDA eingereicht werden müssen. Welche Dokumente einzureichen sind, legt das erstgenannte Guidance-Dokument fest.
Beide HF-Leitlinien sind bei (fast) allen Zulassungsverfahren relevant: 510(k), De Novo, PMA und HDE.
Für einzelne Produktklassen hat die FDA spezielle Guidance-Dokumente zum Human Factors Engineering veröffentlicht. So gibt es ein Guidance-Dokument für Kombinationsprodukte. Ein Fachartikel erläutert die Anforderungen an die Kombinationsprodukte.
2. Human Factors Engineering: Welche Dokumente einzureichen sind
a) Festlegung der Kategorie
Die HF-Dokumentation hängt vom Risiko des Produkts bzw. der Änderung eines bestehenden Produkts ab:
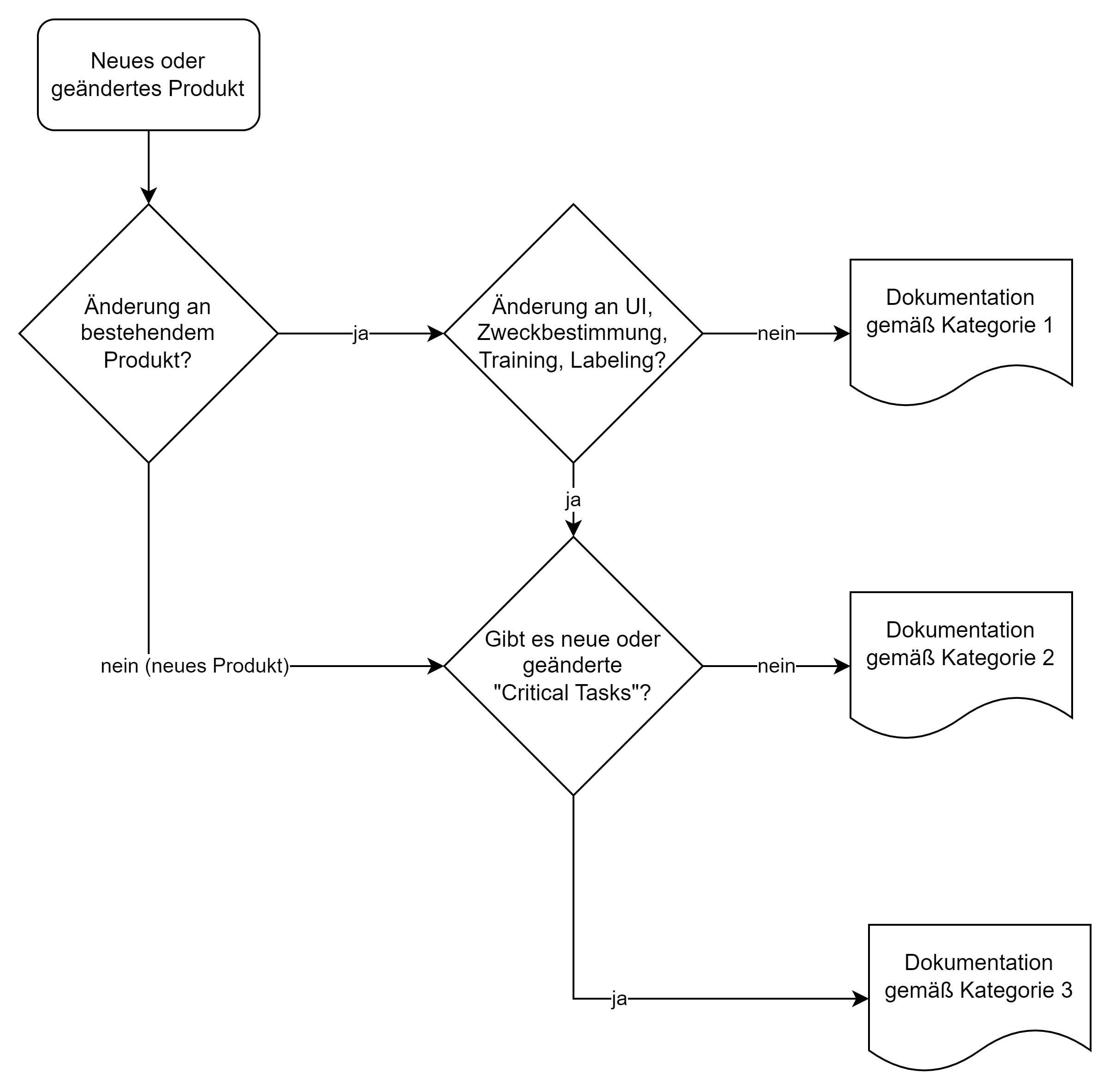
In Kapitel VI (Examples) beschreibt die FDA anhand von Beispielen, wie die jeweiligen Fallunterscheidungen zu treffen sind. Eine Software, bei der die Algorithmen, aber nicht das User Interface geändert wird, wird klassifiziert als eine Änderung, für welche eine Kategorie-I-Dokumentation ausreicht.
b) Festlegung der einzureichenden Unterlagen
Die Kategorien bestimmen den Umfang der einzureichenden HF-Unterlagen:
| Unterlagen | Kat. 1 | Kat. 2 | Kat. 3 |
| Zusammenfassung | X | X | X |
| Beschreibung der Zweckbestimmung (inkl. Nutzer, Nutzungskontext, Training) | X | X | |
| Beschreibung des User Interface | X | X | |
| Zusammenfassung bekannter Nutzungsprobleme | X | X | |
| Zusammenfassung der vorläufigen Evaluationen | X | ||
| Risikoanalyse (Fokus Usability) (inkl. Gefährdungen, Risiken und Beschreibung der „Critical Tasks“) | X | ||
| Details der Usability Validierung | X |
Im Abschnitt „Recommended content of human factors information in marketing submissions“ beschreibt die FDA die in der Tabelle aufgeführten Dokumente genauer.
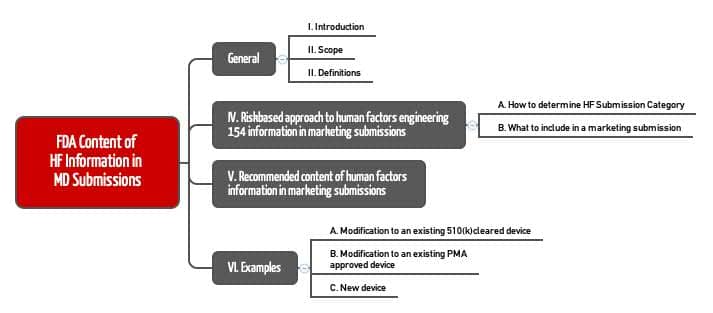
Die „Critical Tasks“ entsprechen weitgehend den „Hazard Relevant Use Scenarios“ der IEC 62366-1. Die FDA definiert „tasks“ inzwischen als „One or more user interactions with a medical device to achieve a desired result“.
Die FDA wünscht sich ein klares Statement zum Vorhandensein oder auch Fehlen von Critical Tasks sowie eine Beschreibung und Bewertung des Restrisikos. Dies kann den Zulassungsprozess der FDA beschleunigen, da diese Informationen nicht nachgefordert werden müssen.
Eine kommentierte Version des FDA-Dokuments zu den einzureichenden Dokumenten können Sie hier herunterladen.
3. Human Factors Engineering: Der HFE-Prozess
a) Das Guidance-Dokument
Das Dokument “Applying Human Factors and Usability Engineering to Medical Devices” der FDA umfasst auf 49 Seiten zehn Kapitel und vier Anhänge.
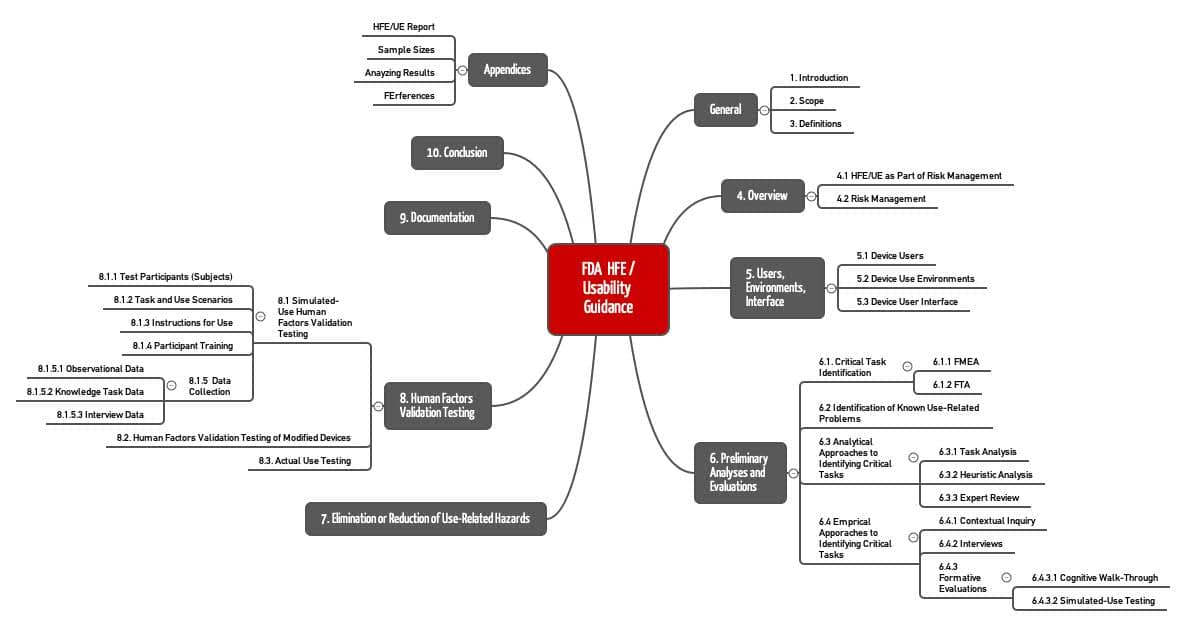
Dieses Dokument enthält in den Kapiteln I bis IV Definitionen und eine Einführung in das Thema. Die FDA beschreibt Konzepte wie Risk Management, User, Use Environment und User Interface.
Weiterhin werden die Aktivitäten beschrieben, die Hersteller im Rahmen des Usability Engineerings durchlaufen sollten:
- Risikoanalyse und Identifizierung kritischer Tasks (z. B. mit FMEA, Tasks-Analyse, Analyse bekannter Nutzungsprobleme, heuristische Evaluation, Expert Review)
- Formative Evaluation (z. T. mit vergleichbaren Methoden)
- Beseitigung von nutzungsbezogenen Gefährdungen
- HF Validation Testing
Die Anforderungen der IEC 62366-1 gleichen sehr den Anforderungen in diesem Guidance-Dokument, auch wenn die Terminologie nicht ganz übereinstimmt. So spricht die Norm nicht von Human Factors Validation Testing, sondern von Summative Evaluation.
Im Gegensatz zur IEC 62366-1 geht die FDA kaum auf den Prozess ein. Hingegen beschreibt sie im HFE-Guidance detailliert die Aktivitäten und Methoden. Letztere sind im TR IEC 62366-2 in ähnlicher Form dargestellt.
b) Umsetzung
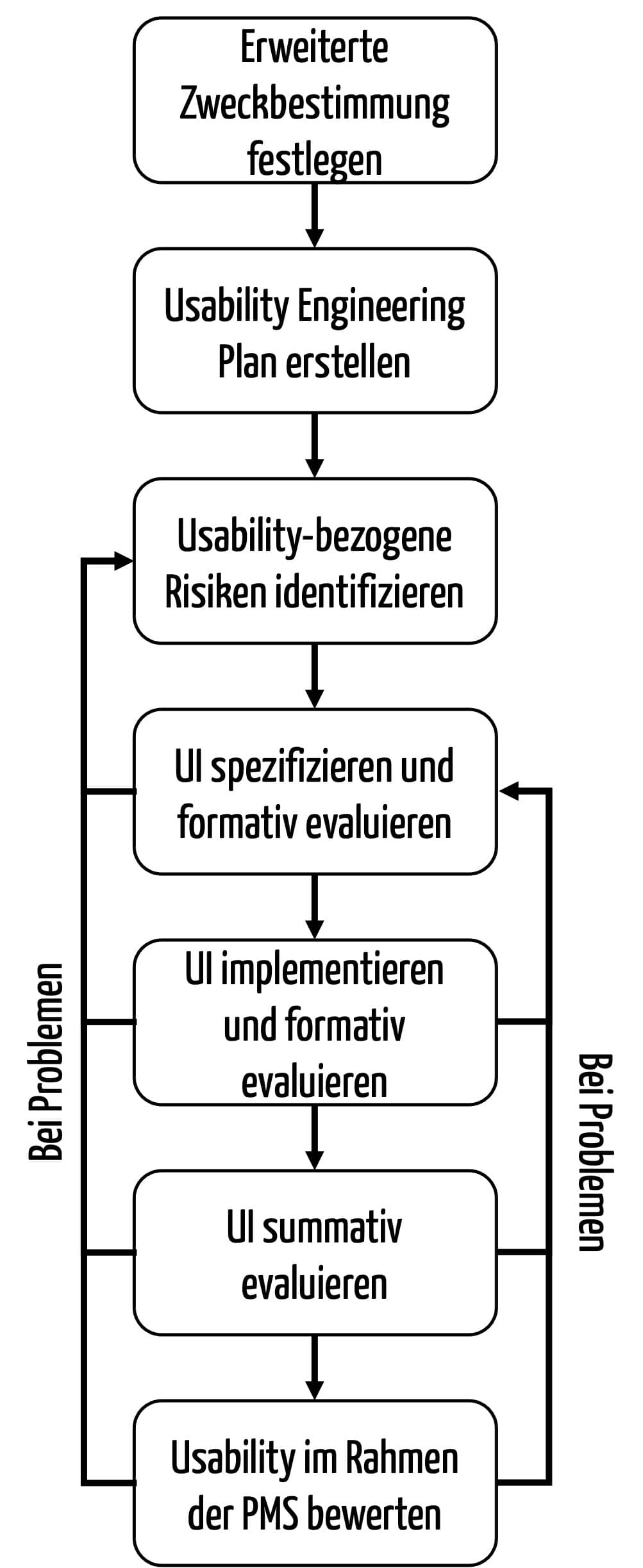
Ein Usability-Engineering-Prozess, der sowohl die Anforderungen der FDA als auch der IEC 62366-1 erfüllt, könnte wie folgt gestaltet sein:
Das Johner Institut unterstützt Medizinproduktehersteller beim FDA-konformen Usability Engineering und hilft, die Zulassungen in allen Teilen der Welt schnell und sicher zu erreichen. Dazu führen unsere Expertinnen und Experten Usability-Studien sowohl in Deutschland und den USA als auch in weiteren Ländern durch.
Lesen Sie hier mehr darüber, wie die Usability Services zur schnellen und sicheren Zulassung Ihrer Produkte beitragen.
4. Zusammenfassung und Fazit
Die Guidance-Dokumente der FDA zum Usability Engineering sind gut verständlich und nachvollziehbar. Der risikobasierte Ansatz beim Zusammenstellen der einzureichenden Dokumentation trägt dazu bei, Zulassungsverfahren schneller durchlaufen zu können, ohne die Sicherheit der Patienten aufgrund mangelnder Gebrauchstauglichkeit zu gefährden.
Wie bei den Vorgaben der FDA zur Software-Dokumentation legt das Guidance-Dokument „Content of Human Factors Information in Medical Device Submissions” die Menge der einzureichenden fest, nicht die der zu erstellenden Dokumentation.
Es ist den Medizinprodukteherstellern sehr dienlich, dass die IEC 62366-1 und die HF-Leitlinien der FDA inzwischen stark abgeglichen sind. Das ermöglicht es beispielsweise, formative und summative Evaluationen so durchzuführen, dass sie die Anforderungen beider Rechtsbereiche erfüllen.
Das Johner Institut hilft gerne, damit die dabei entstehende Dokumentation den regulatorischen Anforderungen genügt.
Änderungshistorie
- 2023-09-15: In Kapitel 1 den Hinweis auf das Guidance-Dokument der FDA zu den Kombinationsprodukten ergänzt
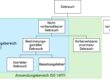

Fehlt in der Abbildung 1 unter 2a nicht die Kategorie 3 (es wird zweimal auf Dokumentation der Kategorie 2 verwiesen)?
Liebe Frau Kleist,
herzlichen Dank fürs aufmerksame Lesen und Ihren wichtigen Hinweis. Sie haben natürlich recht, in der Abbildung muss es korrekterweise Kategorie 3 im letzten Kästchen heißen. Wir werden es direkt korrigieren.
Nochmals vielen Dank für Ihren Kommentar.
Herzliche Grüße
Nils Becker