
Ein ‚Request for Information‘ kann Ihnen teure Anwaltskosten (teilweise) ersparen. Fragen Sie die Behörde doch selbst! So kommen Sie zu einer qualifizierten Antwort. Wenn auch nicht kostenlos.
Lesen Sie hier,
- welche Fragen Ihnen die FDA beantwortet,
- wie Sie vorgehen müssen, um einen ‚Request for Information‘ zu stellen, und
- was die US-Behörde von Ihnen erwartet.
Was ein ‚Request for Information‘ ist
Der US-amerikanische Gesetzgeber verpflichtet seine Behörde – die FDA – dazu, Herstellern von Medizinprodukten innerhalb von 60 Tagen deren Fragen zur Klassifizierung zu beantworten.
Diese Forderung finden Sie im Food, Drug & Cosmetic Act [Artikel 513(g) (§ 360 c.(g) im U.S. Code]:
Within sixty days of the receipt of a written request of any person for information respecting the class in which a device has been classified or the requirements applicable to a device under this Act, the Secretary shall provide such person a written statement of the classification (if any) of such device and the requirements of this Act applicable to the device.
Was sich mit einem ‚Request for Information‘ klären lässt
Klassifizierung
Den ‚Request for Information‘ (RoI) können Sie somit nutzen, um von der FDA eine Aussage darüber zu erhalten, ob Ihr Produkt ein Medizinprodukt ist und, falls ja, in welche generische Produktkategorie und Klasse es fällt.
Zwar muss die Behörde auf die erste Frage (Medizinprodukt ja oder nein) nicht im Rahmen des RoI antworten, sie tut es aber meistens. Andernfalls würden Sie dafür den Request for Designation-Prozess anwenden.
Zulassungsverfahren
Da mit der Klassifizierung häufig die Frage verbunden ist, ob es ein „Substantially Equivalent Device“ gibt, nutzt man den RoI auch, um sich mit der FDA über das Zulassungsverfahren zu verständigen (z. B. 510(k), PMA, IDE, Exempt).
Allerdings möchte die Behörde ihre Einschätzung, ob es ein ausreichend ähnliches Medizinprodukt (Predicate Device) gibt, nicht im Rahmen eines RoI abgeben. Dafür wäre ein Pre-Submission-Meeting die geeignete Wahl.
Weitere regulatorische Voraussetzungen
Neben dem Zulassungsverfahren informiert die Behörde auch über weitere zu beachtende Regularien und Guidance-Dokumente – gegebenenfalls auch über eine ‚Enforcement Discretion‘. Das ist der Verzicht der Behörde, die regulatorischen Forderungen einzufordern.
Wie Sie einen ‚Request for Information‘ stellen
Die FDA beschreibt in dem Guidance-Dokument FDA and Industry Procedures for Section 513(g) Requests for Information under the Federal Food, Drug, and Cosmetic Act, wie Hersteller vorgehen müssen.
1. Schritt: Sich schlau machen
Natürlich wünscht die Behörde, dass man zuerst in den entsprechenden Datenbanken sucht:
Auch soll man entsprechende Veröffentlichungen lesen:
Und schließlich soll man bei speziellen Angelegenheiten Kontakt mit den Spezialisten aufnehmen:
- Kleine Hersteller: Division of Small Manufacturers International and Consumer Assistance: 800-638-2041 oder 301-796-7100 oder DICE@cdrh.fda.gov
- Kombinationsprodukte: Office of Combination Products, 301-796-8930 oder combination@fda.gov
2. Schritt: ‚Request for Information‘ einreichen
Inzwischen ist es möglich, den ROI per eSTAR elektronisch einzureichen. Die passende Vorlage können Sie auf der eSTAR-Webseite der FDA herunterladen.
Alternativ können Hersteller per eCopy-Format einreichen oder ganz klassisch per Post. Wir empfehlen Ihnen allerdings, das zeitgemäße eSTAR-Format zu nutzen.
Ihre Unterlagen sollten die folgenden Informationen beinhalten:
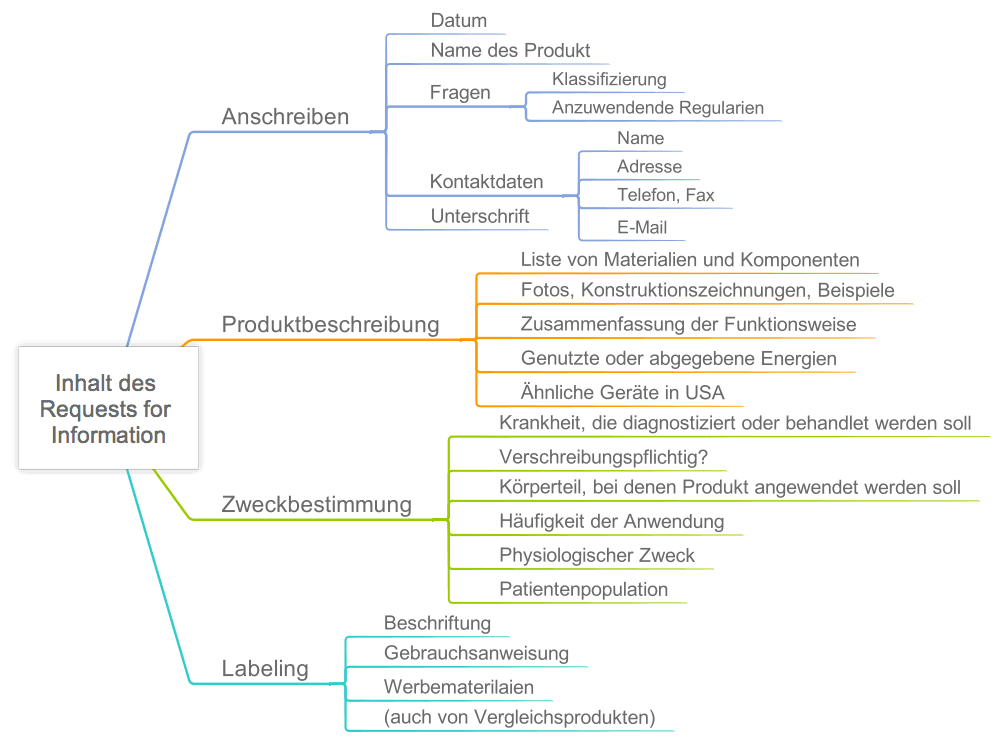
Dem ROI müssen Sie ein „Medical Device User Fee Cover Sheet“ beilegen. Wie Sie das ausfüllen, finden Sie hier beschrieben.
3. Schritt: Gebühren bezahlen
Kostenfrei arbeitet auch die FDA nicht. Über die Gebühren informiert Sie das Guidance-Dokument User Fees for 513(g) Requests for Information. Die Gebühren übersteigen, abhängig von der Firmengröße, die Marke von 7.000 USD. Als „small business“ zahlt man die Hälfte der eigentlichen Gebühr.
Wenn Sie den Antrag eingereicht und die Gebühren bezahlt haben, gibt es keine Chance mehr auf Erstattung – auch nicht, wenn Sie den Antrag zurückziehen. Sie können den Antrag auch nicht durch weitere Fragen ergänzen.
4. Schritt: Rückfragen der Behörde beantworten
Die Behörde kann Rückfragen stellen, die Sie zeitnah beantworten sollten. Eine neue Gebühr fällt nicht an.
5. Schritt: Antwort der Behörde entgegennehmen
Die FDA antwortet schriftlich, im Regelfall innerhalb von 60 Tagen. Das Antwortschreiben ist typischerweise recht knapp und beinhaltet:
- Generische Produktkategorie, falls vorhanden
- Klassifizierung (I, II, III, „unclassified“, „not classified“)
- Zulassungsverfahren (exempt, 510(k), PMA)
- Sonstige zu beachtende Regularien
- Falls vermutlich kein Medizinprodukt vorliegt, die Kontaktinformationen einer anderen Abteilung, welche für diesen Produkttyp zuständig ist.
Falls Sie andere Fragen haben
Konkrete Fragen zur Zulassung Ihres Produkts beantwortet die FDA im Rahmen des ‚Requests for Information‘ nicht. Ziehen Sie hier eine ‚Pre Submission’“‘ in Betracht. Darüber berichten wir im Rahmen eines weiteren Artikels.
Sie möchten wissen, wie Ihr Produkt in den USA zu klassifizieren ist oder benötigen Unterstützung beim Request for Information? Dann nehmen Sie gleich Kontakt auf!
Versionshistorie:
- 2024-11-18: Überarbeitung aufgrund des aktualisierten Guidance-Dokuments. Erwähnung der Einreichung per eSTAR.


