
Die Digitale Gesundheitsanwendungen-Verordnung (DiGAV) ist für die Hersteller von digitalen Gesundheitsanwendungen die wichtigste gesetzliche Vorgabe.
Die DiGAV bestimmt die Voraussetzungen für eine Erstattung von digitalen Gesundheitsanwendungen (DiGA) durch die Krankenkassen.
Erfahren Sie, welche Anforderungen die Verordnung an die Hersteller stellt. So können Sie entscheiden, ob ein Antrag erfolgversprechend ist und ob die Kosten dafür im Verhältnis zum erwarteten wirtschaftlichen Nutzen stehen.
Im April 2020 wurde die Digitale Gesundheitsanwendungen-Verordnung (DiGAV) verabschiedet und im Juni 2021 durch das DVPMG ergänzt.
Parallel zur DiGAV hat das BfArM eine Leitlinie veröffentlicht. Diese verrät auch, welchen amerikanischen Tech-Konzern Sie meiden sollten, um die Anforderungen der DiGAV zu erfüllen. Mehr dazu erfahren Sie im Abschnitt 6 „Anforderungen der DiGAV an die Produkte“.
Sie finden hier die Digitale Gesundheitsanwendungen-Verordnung (DiGAV) und hier die DiGA-Leitlinie des BfArM
1. DiGAV: Um was es geht
Der vollständige Name der DiGAV lautet: „Verordnung über das Verfahren und die Anforderungen zur Prüfung der Erstattungsfähigkeit digitaler Gesundheitsanwendungen in der gesetzlichen Krankenversicherung.“
Diese Verordnung legt fest, wie das „Digitale-Versorgung-Gesetz“ (DVG) umzusetzen ist. Das DVG nennt bereits die wichtigsten Anforderungen an digitale Gesundheitsanwendungen:
- Das Produkt muss ein Medizinprodukt der Klasse I oder IIa (gemäß MDR bzw. MDD) sein.
- Es muss die Anforderungen u.a. an den Datenschutz und die Interoperabilität erfüllen.
- Der Hersteller hat positive Versorgungsaspekte nachzuweisen.
- Der Hersteller muss einen erfolgreichen Antrag beim BfArM zur Aufnahme in das „Verzeichnis erstattungsfähiger digitaler Gesundheitsanwendungen nach § 33a“ stellen.
Im Artikel zum DVG finden Sie eine Übersicht über die Voraussetzungen, die DiGA-Hersteller erfüllen müssen, und über das Antragsverfahren.
Ein weiterer Artikel beschreibt die 7 Schritte, mit denen Hersteller DiGA ins DiGA-Verzeichnis und damit bis zur Erstattungsfähigkeit bekommen.
Die DiGAV beschreibt genauer als das DVG, wie die Hersteller nachweisen können, dass sie bzw. ihre Produkte die gesetzlichen Anforderungen erfüllen. Beispielsweise enthält die Verordnung konkrete Checklisten, anhand derer die Hersteller überprüfen müssen, ob die Anforderungen an die IT-Sicherheit erfüllt sind.
Die Verordnung regelt im Gegensatz zum Gesetz auch die Kosten, den Verfahrensablauf und die genauen Inhalte des elektronischen Verzeichnisses.
2. DiGAV im Überblick
Die DiGAV umfasst 43 Paragrafen, die in 9 Abschnitte gegliedert sind (s. Abb. 1).
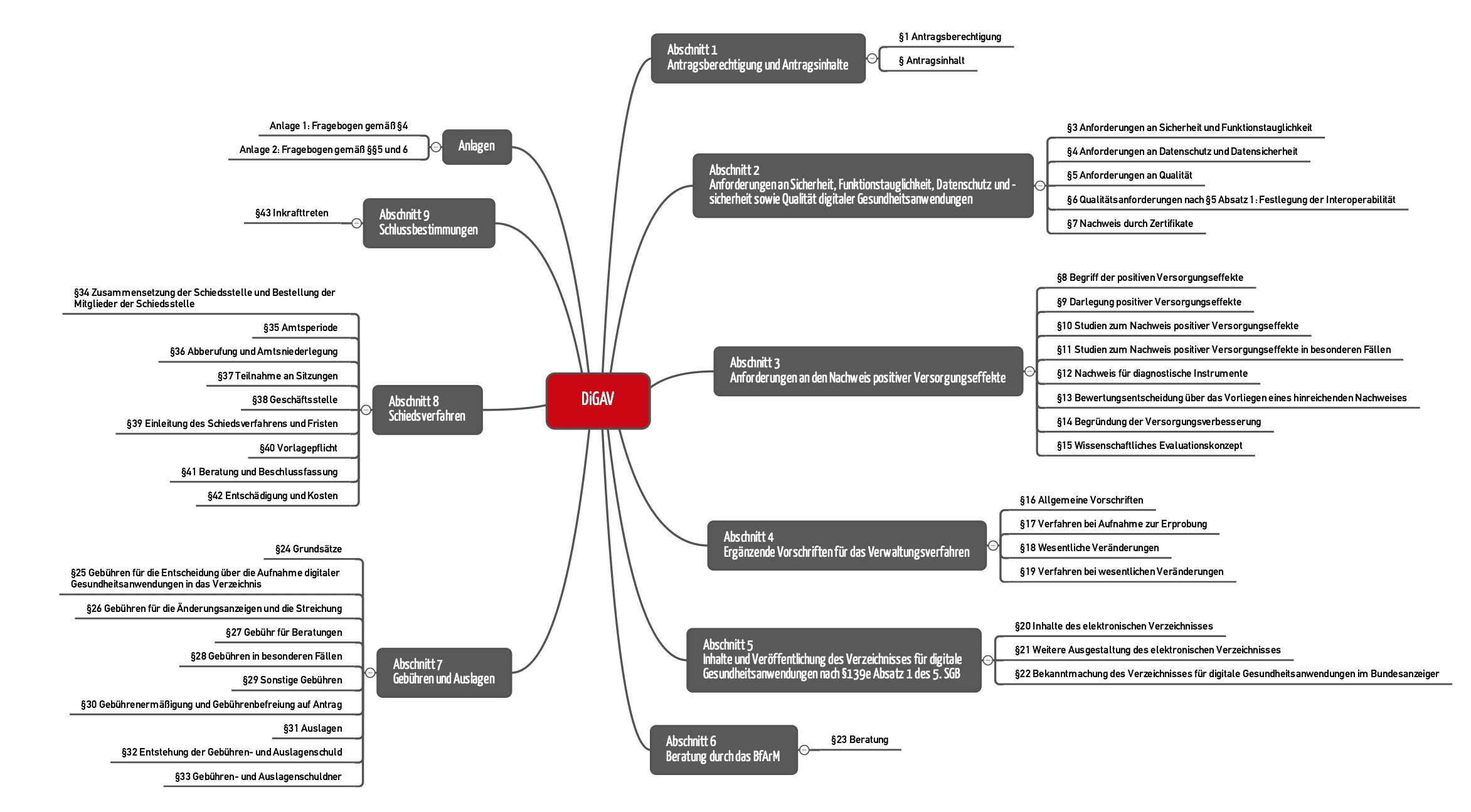
Besonders relevant für die Hersteller sind die Abschnitte 2, 3 und 5:
- Abschnitt 2 legt die Anforderungen an die DiGA fest, z.B. bezüglich Datensicherheit, Interoperabilität und Qualität.
- Abschnitt 3 beschreibt, wie die Hersteller nachweisen müssen, dass ihr Produkt einen „positiven Versorgungsaspekt“ aufweist.
- Diejenigen Inhalte, die Hersteller im „DiGA-Verzeichnis“ öffentlich machen müssen, bestimmt der Abschnitt 5.
Was das Digitale-Versorgung-Gesetz unter einem positiven Versorgungsaspekt versteht und welche Beispiele es gibt, können Sie hier nachlesen.
3. Ab wann man mit DiGA Geld verdienen kann
Die Bundesregierung scheint daran interessiert zu sein, dass Hersteller rasch von der Erstattung ihrer DiGA profitieren können. Das BfArM schreibt: „Das Verfahren ist als zügiger „Fast-Track“ konzipiert.“
Ob „zügig“ nun als Tautologie oder als Relativierung von „Fast“ zu verstehen ist, lässt die Behörde offen.
In seinem Leitfaden zeigt das BfArM jedenfalls auf, dass im August die ersten DiGA ins Verzeichnis aufgenommen sein können.
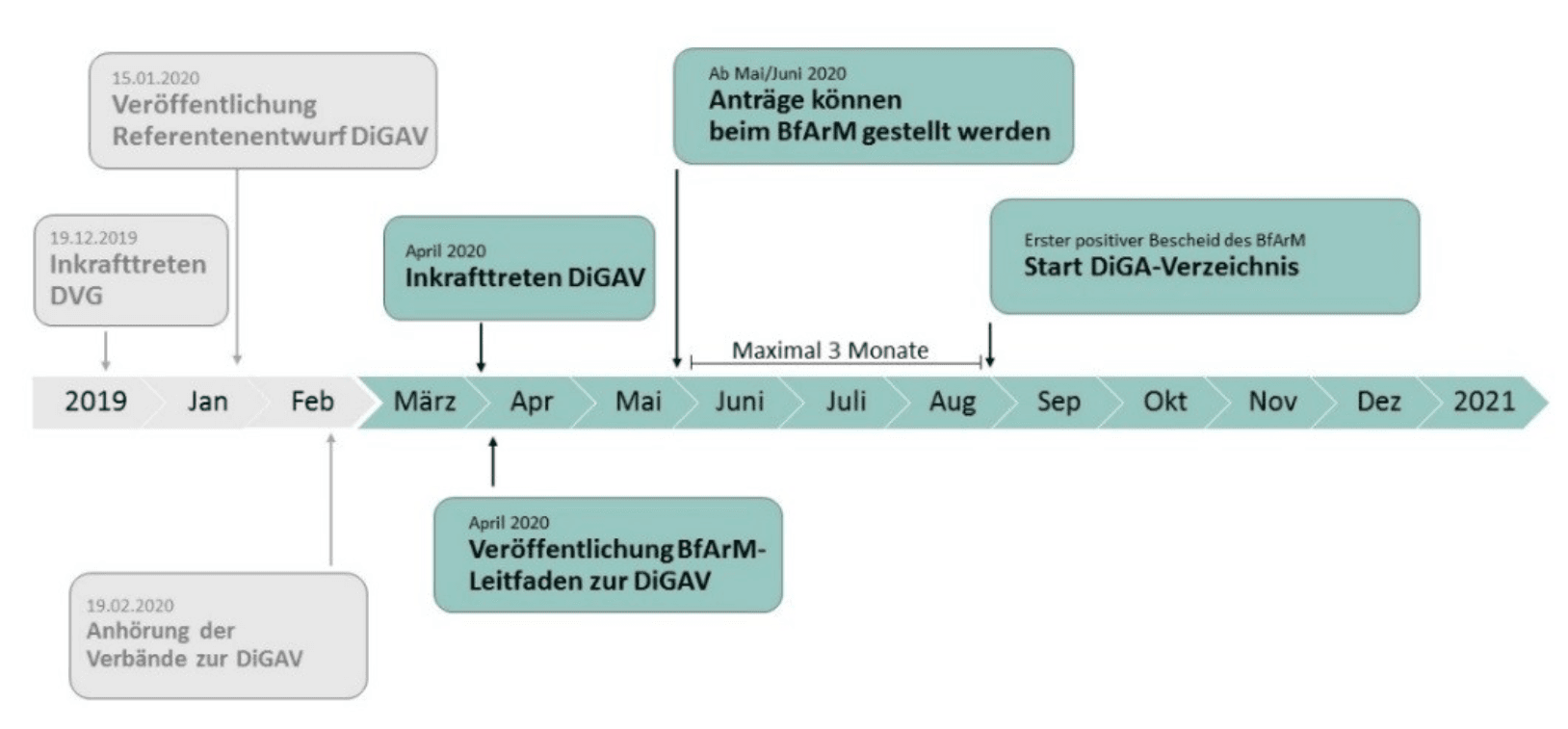
Das Fast-Track-Verfahren hat sich bewährt. Innerhalb von drei Monaten erreichen die Hersteller in der Regel eine Entscheidung: Annahme, Ablehnung oder Zurückziehen.
Beim Zurückziehen, darf man jederzeit einen neuen Versuch wagen, bei der Ablehnung muss man ein Jahr warten bis zum nächsten Antrag.
4. Was die Antragsstellung kostet (§§ 24 ff.)
Die DiGAV benennt die Kosten für eine Antragsstellung. Diese belaufen sich typischerweise auf 3.000 bis 9.900 EUR.
- 3.000 bis 9.900 EUR für die Entscheidung über die dauerhafte Aufnahme in das Verzeichnis für digitale Gesundheitsanwendungen nach § 139e Absatz 3 Satz 1 und für die Entscheidung über die vorläufige Aufnahme in das Verzeichnis für digitale Gesundheitsanwendungen zur Erprobung nach § 139e Absatz 4 Satz 1 und 3
- 1.500 bis 6.600 EUR für die Entscheidung über die dauerhafte Aufnahme in das Verzeichnis nach Abschluss der Erprobung nach § 139e Absatz 4 Satz 6
- 1.500 bis 4.900 EUR für die Entscheidung über die Verlängerung der Erprobungszeit nach § 139e Absatz 4 Satz 7.
Beratungen kosten zwischen 250 und 5.000 EUR, wobei „im Umfang geringfügige allgemeine mündliche, schriftliche oder elektronische Auskünfte hiervon ausgenommen sind“.
Auf Antrag darf das BfArM die Gebühren bis auf ein Viertel reduzieren, beispielsweise wenn der Hersteller keinen angemessenen wirtschaftlichen Nutzen erwarten kann. Das wäre bei sehr kleinen Zielgruppen der Fall.
5. Verzeichnis für digitale Gesundheitsanwendungen (§§ 20 ff.)
5.1 Inhalte des Verzeichnisses
Die DiGAV beschreibt im § 20, welche Inhalte im Verzeichnis für digitale Gesundheitsanwendungen veröffentlicht werden müssen. Eine noch genauere Vorgabe findet sich im Leitfaden des BfArM, Kapitel 2.2 (s. Abbildung 3).
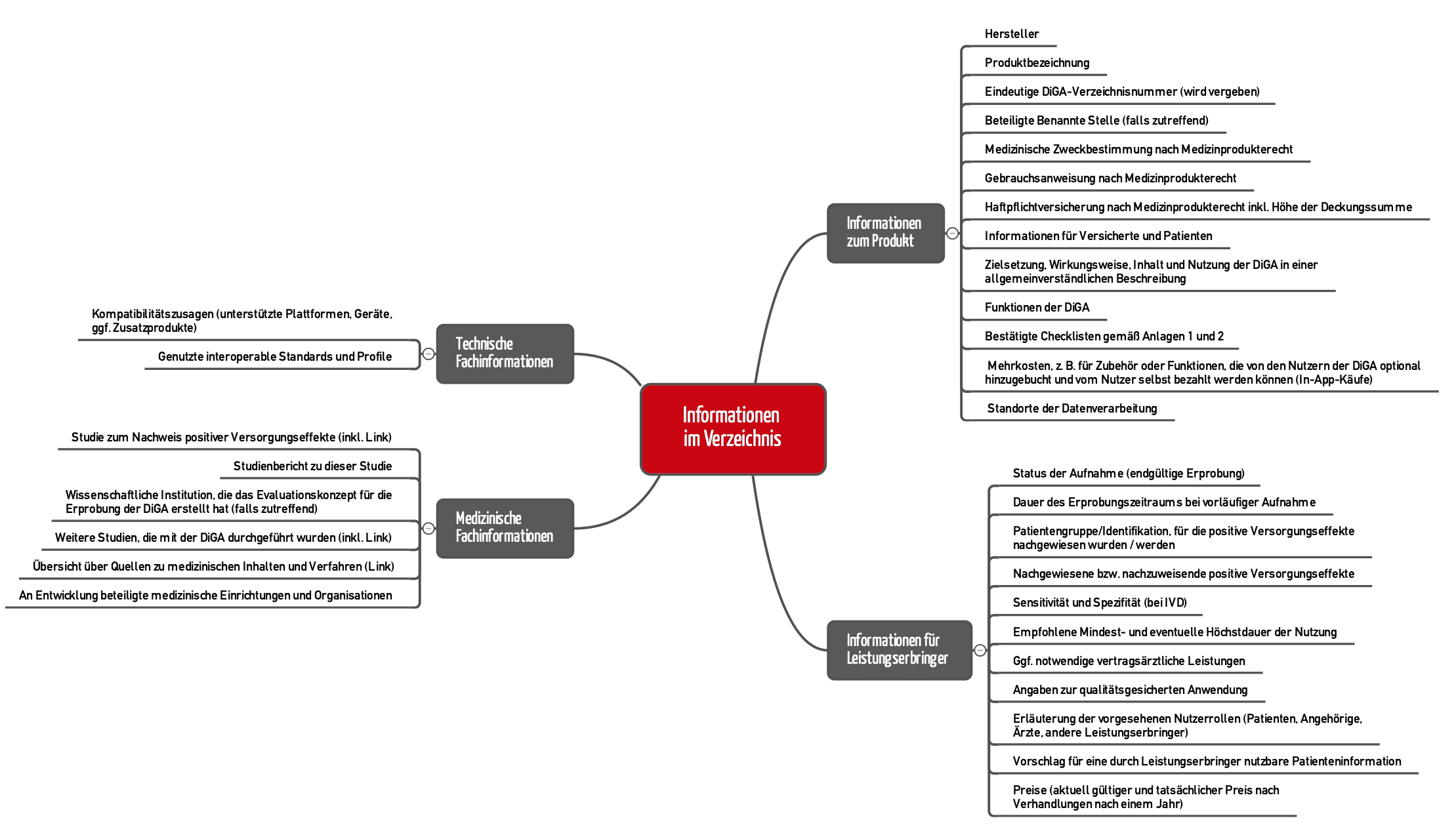
In einigen Fällen genügt es jedoch, einen Link auf eigene Webseiten zu hinterlegen, auf denen die Informationen zu finden sind.
Die Hersteller müssen sich damit ziemlich entblößen: Gerade die Pflicht, auch die Studien und ihre Ergebnisse zu veröffentlichen, dürfte vielen aufstoßen. Mancher Hersteller mag befürchten, dass damit Geschäftsgeheimnisse offenbart werden müssen. Das BfArM schreibt dazu:
Der Hersteller kann im Antragsverfahren die Angaben kennzeichnen, bei denen rechtliche Anforderungen einer Veröffentlichung entgegenstehen. Beispiele hierfür sind der Schutz von Betriebs- und Geschäftsgeheimnissen, der Schutz personenbezogener Daten Dritter oder der Schutz von geistigem Eigentum.
DiGA-Leitfaden des BfArM
Das BfArM besteht aber in jedem Fall auf einem vollständigen Antrag.
5.2 Schnittstellen des Verzeichnisses
Das Verzeichnis soll öffentlich zugänglich sein. Es sind zwei Schnittstellen vorgesehen:
- „Nutzerfreundliches und zielgruppenspezifisch strukturiertes Webportal“
- Programmierschnittstelle (Application Programming Interface, API)
Das Webportal soll „Versicherten oder Ärzten in unterschiedlichen Ansichten die für sie besonders relevanten Informationen in übersichtlicher Darstellung bereitstellen“.
Die API soll „anderen interessierten öffentlichen und gemeinnützigen Institutionen“ zur Verfügung gestellt werden. Das BfArM denkt dabei an „Fachgesellschaften, Krankenkassen, Ärzteverbände, Forschungsinstitutionen, Stiftungen, Kommunen, Patientenverbände und weitere Akteure“.
Diese API ist allerdings noch nicht spezifiziert. „Details zu der Programmierschnittstelle und deren Nutzung (Registrierung, Testzugänge etc.) wird das BfArM im Laufe des Jahres über seine Website veröffentlichen.“
6. Anforderungen der DiGAV an die Produkte
6.1 Datensicherheit und Datenschutz
Eine Übersicht über alle regulatorischen Anforderungen finden Sie im Artikel zur Datensicherheit und dem Datenschutz bei DiGA.
Die DiGAV konkretisiert die Anforderungen (zum Glück), indem sie in Anlage 1 Checklisten anbietet. Leider scheinen diese Checklisten nur bedingt mit dem BSI TR-03161 des BSI abgestimmt zu sein.
Die Anlage enthält auch weiterhin nur bedingt sinnvolle Anforderungen, z.B. XML oder JSON-Dateien gegen definierte Schemata zu prüfen, um DDoS-Angriffe abzuwehren.
Das kann helfen, es kann aber auch genau das Gegenteil bewirken: Wer alle Nachrichten eines DDoS-Angriffs erst gegen Schemata prüft, wird seinen Server noch schneller in die Knie zwingen.
Unterscheidung von hohen und sehr hohen Schutzanforderungen
Die Checkliste der DiGAV enthält Punkte, die nur bei DiGA mit „sehr hohem Schutzbedarf“ zu beachten sind. Beispielsweise besteht die Verordnung nur bei sehr hohem Schutzbedarf auf Penetrationstests oder einer 2-Faktor-Authentifizierung.
Überlassen Sie die Sicherheit Ihrer Patienten nicht dem Zufall
Gehen Sie mit einem Pentest des Johner Instituts auf Nummer sicher!
Was ein sehr hoher Schutzbedarf ist, definiert das BSI im BS 200-2 auf Seite 24. So liegt ein sehr hoher Schutzbedarf vor, wenn „der Schutz personenbezogener Daten unbedingt gewährleistet sein muss. Anderenfalls kann es zu einer Gefahr für Leib und Leben oder für die persönliche Freiheit des Betroffenen kommen.“
Daten auf den Servern von Amazon
Nicht die DiGAV, sondern der Leitfaden des BfArM ringt sich zu halbwegs konkreten Aussagen durch, ob Hersteller die Cloud-Dienste amerikanischer Tech-Giganten nutzen dürfen:
Da § 4 Absatz 3 DiGAV grundsätzlich nur die Verarbeitung von personenbezogenen Daten in Drittstaaten zulässt, wenn ein Angemessenheitsbeschluss nach Art. 45 DSGVO vorliegt, ist eine Verarbeitung von personenbezogenen Daten in den USA nicht zulässig. Eine Lösung über Standardvertragsklauseln ist nicht erlaubt. Weiterhin ist eine Datenverarbeitung in den USA nach Patienteneinwilligung gemäß Art. 49 DSGVO ebenfalls nicht zulässig, da nach Verordnung einer DiGA durch einen Arzt die Freiwilligkeit des Patienten nicht mehr gegeben ist.
DiGA-Leitfaden des BfArM S. 46
Damit dürfen personenbezogene Daten nicht in der Apple Cloud gespeichert werden. Wer glaubt, man könne einfach auf Amazon ausweichen, ggf. in einem deutschen Rechenzentrum, sollte das Dokument weiterlesen:
Dienstleister aus den USA, auch solche mit Niederlassung in der EU, aber einem Mutterkonzern in den USA, dürfen aufgrund des EuGH-Urteils und den Vorgaben der DiGAV nicht für die Verarbeitung von personenbezogenen Daten herangezogen werden. Auch für jegliche Tools, die im Rahmen der Nutzung der DiGA zum Einsatz kommen, muss ein Datenfluss von personenbezogenen Daten in die USA vollumfassend ausgeschlossen werden.
DiGA-Leitfaden des BfArM S. 46
Diese Vorgaben werden viele DIGA-Hersteller vor Probleme stellen. Zumindest sind sie eindeutig.
6.2 Interoperabilität
Ziel der Interoperabilität
Auch die Interoperabilität der DiGA liegt dem Gesetzgeber am Herzen. Das hat mehrere Gründe:
- Patienten sollen Versicherungen wechseln und dabei ihre Daten mitnehmen können. Andernfalls wäre ein Wettbewerb zwischen den Versicherungen erschwert.
- Die Gesundheitsversorgung muss sektorenübergreifend gelingen. Sonst fallen unnötige Kosten an, z.B. für Doppeluntersuchungen, für die Übertragung von einem System in ein anderes, für manuelle Tätigkeiten usw.
Die Interoperabilität soll den durchgängigen Informationsfluss zwischen allen am Gesundheitswesen Beteiligten und deren IT-Systemen und (Medizin-) Geräten ermöglichen. Dazu gehören:
- Krankenhäuser
- Niedergelassene Ärzte
- Apotheken
- Therapeuten
- Krankenkassen
- Patienten (inkl. deren eigene Messgeräte, auch Wearables)
Daher formuliert die DiGAV genaue Anforderungen an die Interoperabilität.
„Ausgezeichnete Interoperabilitätsstandards“ verwenden
Die Hersteller sind nicht frei in der Wahl der Interoperabilitätsstandards.
- Sie müssen ein von der KBV definiertes MIO oder einen im Vesta-Verzeichnis (Verzeichnis für informationstechnische Standards im Gewundheitswesen, siehe Hinweis unten) als empfohlen ausgezeichneten Standard oder ein ebensolches Profil umsetzen.
- Falls es dort keinen passenden Standard gibt, muss der Hersteller eine der folgenden Optionen wählen:
- Existierende offene, international anerkannte Standards nutzen, z. B. ein von HL7 definiertes FHIR-Profil
- Eigenes Profil für einen bestehenden offenen, internationalen Standard entwickeln bzw. ein Profil erweitern. Das setzt aber voraus, dass „der Hersteller bei der gematik die Aufnahme der so entstandenen Schnittstellenspezifikation im vesta Verzeichnis“ beantragt.
- Eigenes Profil für einen im vesta-Verzeichnis gelisteten Standard weiterentwickeln bzw. ein Profil erweitern. Das setzt ebenfalls voraus, dass der Hersteller „bei der gematik die Aufnahme der so entstandenen Schnittstellenspezifikation ins vesta Verzeichnis“ beantragt.
Entweder Sie nutzen einen Standard (wenn es einen gibt) oder Sie tragen dazu bei, einen Standard zu erweitern oder neu zu schaffen.
Auf Basis der Gesundheits-IT-Interoperabilitäts-Governance-Verordnung (GIGV) und der im Oktober 2021 in Kraft getretenen Rechtsverordnung der GIGV wurde aus dem Vesta-Verzeichnis der Interoperabilitäts-Navigator INA für digitale Medizin (Quelle).
Ausgezeichnete Interoperabilitätsstandards auch bei Medizingeräten
Diese Pflicht zur Verwendung von ausgezeichneten Standards findet sich auch bei Medizingeräten, Wearables und Sensoren. Die DiGAV stellt den Herstellern drei Optionen zur Verfügung.
- Sie implementieren „ein offengelegtes und dokumentiertes Profil des ISO/IEEE 11073 Standards“.
- Sie verwenden einen im „vesta Verzeichnis verzeichneten Standard bzw. ein dort verzeichnetes Profil“.
- Der Hersteller entwickelt ein „eigenes Profil bzw. einen eigenen Standard und beantragt die Aufnahme dieser Spezifikation im vesta Verzeichnis“.
Diese Forderungen gelten aber nur, wenn der Hersteller die Daten direkt von einem Gerät und nicht nur indirekt über eine Plattform wie Apples Health Kit bezieht.
Dass das BfArM ausgerechnet Apple Health erwähnt, überrascht. Denn die Behörde hat doch selbst festgestellt, dass Apple nicht unter den Privacy Shield fällt.
Fazit: Die DiGAV treibt die „Standardisierung der Standards“ voran, was zu begrüßen ist. Dies wird das Ende vieler proprietärer Standards sein.
Welche Daten ausgetauscht werden müssen
Die Hersteller müssen nicht alle Daten über die standardisierten Schnittstellen anbieten. Beispielsweise dürften Log-Dateien, Rohdaten oder Nutzungsstatistiken auch über proprietäre Schnittstellen „ausgespielt“ werden.
Das BfArM formuliert die folgende Faustregel:
Die Forderung nach einem interoperablen, maschinenlesbaren Export ist ausschließlich eine Anforderung an die Interoperabilität. Interoperabilität geht vor Vollständigkeit. Wenn ein MIO oder ein im vesta Verzeichnis empfohlener Standard/Profil/Leitfaden bekannt ist, worüber sich 80 Prozent der eigentlich zu exportierenden Inhalte abdecken lassen, dann muss dieser verwendet werden.
DiGA-Leitfaden des BfArM
6.3 Robustheit
Die DiGAV und der Leitfaden formulieren noch klarer als das DVG, was der Gesetzgeber unter „Robustheit“ versteht: Es geht um die Fähigkeit des Systems, mit zum Beispiel folgenden Ereignissen umgehen zu können:
- Ausfall der Stromversorgung
- Störung der Internetverbindung
- Abschalten des Mobilgeräts
- (Versehentliches) Entkoppeln eines Geräts
- Falsche und unvollständige Systemeinstellungen
- Problem mit Hardware, Sensorik, Aktoren
- Fehleingaben durch Benutzer
Der BfArM-Leitfaden nennt auch Konsequenzen, die bei den o.g. Ereignissen vermieden werden müssen:
- Verfälschung von Daten
- Verlust von Daten
- Falsche (unwahrscheinliche, unmögliche) Daten
- Daten mehrfach vorhanden
- System in inkonsistentem Zustand
- Unnötige Aufwände für Anwender
Selbst mögliche Maßnahmen nennt der Leitfaden:
- Prüfung von Daten auf Plausibilität
- Verwerfen falscher Daten
- Hinweise bzw. Warnungen an Benutzer
- Nutzen von Referenz- bzw. Testdaten (z.B. Bilder)
- Möglichkeit eines Resets
6.4 Nutzerfreundlichkeit (§ 5 Absätze 5 und 6)
Leider arbeiten die DiGAV und das BfArM mit dem nicht definierten Begriff „Nutzerfreundlichkeit“. Es ist nicht klar, ob damit die Gebrauchstauglichkeit gemeint ist oder eher die Benutzbarkeit.
Zumindest ein starker Fokus auf die Barrierefreiheit lässt vermuten, dass es um die Benutzbarkeit geht. Die DiGAV unterscheidet in der Anlage 2 allerdings zwischen den beiden Begriffen.
Zur Umsetzung der Barrierefreiheit kann der Hersteller sich an Vorgaben und Hinweisen von Organisationen wie beispielsweise der Bundesfachstelle Barrierefreiheit orientieren. Die Umsetzung der Barrierefreiheit ist immer zielgruppen- und nutzungsspezifisch zu prüfen.
Hinweise im Leitfaden auf „Styleguides“ und auf die „Durchführung von Fokusgruppen“ deuten Richtung Gebrauchstauglichkeit im weiteren Sinn. Diese müsste im Rahmen der „Zulassung“ als Medizinprodukt aber bereits nachgewiesen sein.
Unser Usabiltiy Team des Johner Instituts unterstützen Sie gerne bei den Usability-Tests und deren Dokumentation. Dabei verknüpfen sie die Erstellung der Gebrauchstauglichkeitsakte mit der Erfüllung der Anforderungen an Benutzerfreundlichkeit und Barrierefreiheit. Nehmen Sie Kontakt auf z.B. über das Kontaktformular.
6.5 Qualitätsgesichertes Wissen (§ 5 Absatz 8)
Die DiGAV fordert:
Die von digitalen Gesundheitsanwendungen verwendeten medizinischen Inhalte müssen dem allgemein anerkannten Stand der medizinischen Erkenntnisse entsprechen.
DiGAV § 5 (8)
Das BfArM schließt daraus, dass „sich die medizinisch-fachliche Grundlage der DiGA aus akzeptierten und belastbaren Quellen wie z.B. medizinischen Leitlinien, etablierten Lehrbüchern, vergleichbaren anerkannten Quellen oder zumindest veröffentlichten Studien ableiten muss.“
6.5 Ergänzte Anforderungen an die DiGA durch das DVPMG
Im Juni 2021 wurde das DVPMG (Digitale-Versorgung-und-Pflege-Modernisierungs-Gesetz) erlassen. Dieses Gesetz ändert u. a. das SGB V sowie die DiGAV. Eine Übersicht über diese Änderungen finden Sie im Kapitel 4 des Artikels zum DVPMG.
7. Nachweis der positiven Versorgungsaspekte
Wahrscheinlich immer noch die größten Bauchschmerzen dürfte den Herstellern der Abschnitt 3 „Anforderungen an den Nachweis positiver Versorgungseffekte“ bereiten.
Die Definitionen der Begriffe „positiver Versorgungseffekt“, „medizinischer Nutzen“ und „patientenrelevante Struktur- und Verfahrensverbesserungen“ sowie Beispiele dafür finden sich im Beitrag zum DVG, auch wenn erst die DiGAV bzw. das BfArM diese Beispiele nennt.
Hersteller aller Medizinprodukte müssen nachweisen, dass ihre Produkte zumindest den Stand der Technik reflektieren und damit einem alternativen Produkt oder Verfahren gleichwertig sind.
Die DiGAV legt die Latte höher, wie Sie im Folgenden ausgeführt finden.
Herausforderung 1: Die DiGA muss besser sein
Der Hersteller muss nachweisen, dass die DiGA besser(!) ist als deren Nichtanwendung. Dabei versteht man unter Nichtanwendung, dass ein Patient
- ohne die entsprechende DiGA, aber mit etwas anderem behandelt wird,
- gar nicht behandelt wird oder
- mit einer vergleichbaren und bereits gelisteten DiGA behandelt wird.
In der Praxis muss der medizinische Nutzen immer im Vergleich zur Standardbehandlung / Regelversorgung nachgewiesen werden.
Das hat zur Folge, dass es nicht möglich sein wird, eine gleichwertige DiGA in das Verzeichnis der DiGA-Anwendungen aufnehmen zu lassen. Für Hersteller bedeutet das, dass der Erste gewinnt!?
Es ist daher nicht ganz nachvollziehbar, wie eine Situation auftreten kann, die das BfArM hier schildert:
Sofern für den verordnenden Arzt in Bezug auf die angestrebte Therapieunterstützung im konkreten Behandlungsfall kein Unterschied zwischen zwei oder mehr DiGA erkennbar ist, kann es im Einzelfall geboten sein, die kostengünstigere DiGA zu verordnen.
Diga-Leitfaden des BfArM
Herausforderung 2: Der Nachweis muss in Form einer Studie erfolgen
Die MDR bzw. MDD erlauben es, im Rahmen der klinischen Bewertung den Nachweis des Nutzen-Risiko-Verhältnisses anhand von Literatur und ggf. sogar „nur“ anhand von Leistungsdaten zu führen.
Die DiGAV hingegen besteht immer auf einer wissenschaftlichen Studie.
Herausforderung 3: Der Studie muss mit der DiGA selbst durchgeführt werden
Auch bei Produkten, mit denen die Nachweise im Rahmen der Studien erbracht werden, zeigen sich MDR und MDD großzügiger: Solange das Äquivalenzprodukt klinisch, technisch und biologisch vergleichbar ist, dürfen damit gewonnene Daten zum Nachweis genutzt werden.
Die DiGAV geht einen Schritt weiter. Das BfArM umschreibt die Forderung wie folgt:
Alleinige Verweise auf andere Primärliteratur und Studien auch von anderen gleichartigen DiGA sind nicht zulässig.
DiGA-Leitfaden des BfArM
Herausforderung 4: Die Studien müssen in Deutschland durchgeführt werden
Das BfArM kommt zu dem Schluss: „Durch die Begrenzung auf Deutschland ist gewährleistet, dass die Studienergebnisse ausreichend aussagekräftig sind.“
Dieser Schlussfolgerung kann das Johner Institut nicht folgen: Gerade durch die Begrenzung auf Deutschland kann es vorkommen, dass nicht ausreichend Daten gesammelt werden können und die Studienergebnisse gerade nicht ausreichend aussagekräftig sind.
Ob Studienergebnisse aussagekräftig sind, hängt von Art und Güte der Studie und von der Vergleichbarkeit der Population ab, aber nicht primär vom Land.
Würden andere Länder solche Forderungen erheben, würde man das als Wettbewerbsbeschränkung brandmarken. Dass also gegen diesen Punkt der Verordnung geklagt werden wird, halten wir für wahrscheinlich.
Herausforderung 5: Veröffentlichungspflicht
In der DiGAV ist vorgeschrieben, dass die Studien registriert und deren Ergebnissen publiziert werden. Das BfArM schreibt:
Die Studien müssen in einem öffentlichen Studienregister registriert werden. Das Studienregister muss ein Primärregister oder ein Partnerregister der World Health Organisation International Clinical Trials Registry Platform oder ein Datenlieferant der World Health Organisation International Clinical Trials Registry Platform sein. […] Das anerkannte Primärregister für Deutschland ist das Deutsche Register klinischer Studien (DRKS) beim Deutschen Institut für Medizinische Dokumentation und Information (DIMDI).
DiGA-Leitfaden des BfArM
Studien, die für die Aufnahme ins Verzeichnis vorgelegt werden, [müssen] spätestens zwölf Monate nach Studienabschluss vollständig mit den Ergebnissen im Internet veröffentlicht werden. […]
Wichtig ist, dass auch negative Ergebnisse veröffentlicht werden.
Diese Informationen sind auch für die Konkurrenz interessant. Wenn aus den Ergebnissen Rückschlüsse auf Geschäfts- und Betriebsgeheimnisse gezogen werden können, dürfen entsprechende Stellen geschwärzt werden.
8. Fazit
Zusammenfassung
Für die Hersteller mag es verlockend klingen, ihre DiGA von den Krankenkassen erstattet zu bekommen. Doch die DiGAV legt die Latte dafür sehr hoch:
- Die Hersteller müssen die Anforderungen an den Datenschutz anhand einer Checkliste nachweisen.
- Ihre Produkte müssen die Interoperabilität i.d.R. durch allgemein anerkannte Standards gewährleisten. Jemand, der noch nie von MIOs oder einer vesta-Liste gehört hat, wird sich damit sicher schwertun.
- Die Pflicht zur Durchführung von Studien mit der eigenen DiGA sowie die Pflicht, besser als alternative Produkte oder Verfahren zu sein, bedeutet für die Hersteller hohe Aufwände. Die Anforderungen durch die DiGAV sind höher als die Anforderung der MDR an die klinische Bewertung. Doch bereits bei der MDR klagen die Hersteller über die Aufwände.
Zustimmung und Kritik
Wer Geld von der Gesellschaft, sprich den Versicherten will, muss etwas leisten. Das ist gut so. Es ist zu befürworten, dass
- relativ präzise Anforderungen an den Datenschutz gestellt werden,
- der Zwang zu „standardisierten Standards“ zum Einreißen der Sektorengrenzen im Gesundheitswesen führen und damit dessen Effizienz (und hoffentlich auch Effektivität) erhöhen wird,
- der Nutzen nachgewiesen sein muss und dass für Anwendungen mit fraglichem Nutzen kein öffentliches Geld ausgegeben wird – zumindest nicht dauerhaft.
Es bleiben aber Fragen offen:
- Aus welchem Grund werden Produkte der Klassen IIb und III ausgeschlossen, die oft einen besonders hohen Nutzen haben?
- Weshalb sind die Checklisten-Punkte zur IT-Sicherheit in der Anlage I der DiGAV nicht mit dem BSI TR-03161 abgeglichen?
- Weshalb glaubt man, dass die Aussagekraft von Studien höher ist, wenn diese nur in Deutschland durchgeführt werden? Dass sich die DiGAs in der deutschen Versorgungsrealität beweisen müssen, ist hingegen verständlich.
- Wie soll ein Wettbewerb zwischen DiGA möglich sein, wenn es nicht erlaubt ist, eine „gleich gute“ DiGA ins Verzeichnis aufzunehmen?
- Weshalb muss ein Hersteller besser als der Stand der Technik sein? Hersteller von Medizinprodukten müssen den medizinischen Nutzen bereits nachgewiesen haben. Der Grund liegt wahrscheinlich darin, dass die Kosten sonst nicht vertretbar sind.
- Weshalb besteht die DiGAV auch bei Produkten, die nur einen medizinischen Nutzen und keine sonstigen positiven Versorgungsaspekte in Anspruch nehmen, auf neuen Studien?
Dass die Hürden so hoch gelegt wurden, ist eine politische Entscheidung, die mit Blick auf die Sicherheit von Patienten und sinnvolle Nutzung öffentliche Gelder nachvollziehbar ist.
Überraschenderweise hat die Praxis gezeigt, dass die Startups die Nase vorn haben, obwohl die Anforderungen der Digitale Gesundheitsanwendungen-Verordnung immens sind und on top zu den MDR-Anforderungen kommen. Die großen Firmen verfügen zwar über mehr Geld, tun sich aber dennoch schwer damit.
Aktuelles
Änderung der Digitale Gesundheitsanwendungen-Verordnung (DiGAVÄndV)
Mit der ersten Verordnung zur Änderung der Digitale Gesundheitsanwendungen-Verordnung (DiGAVÄndV) ergeben sich für die Hersteller von Digitalen Gesundsheitsanwendungen (DiGA) einige Neuerungen:
Zusammenfassung
Die wichtigsten Erkenntnisse aus der Änderung für Sie als Hersteller:
- Der Antrag und die Angaben für das DiGA-Verzeichnis müssen in deutscher Sprache vorliegen
- Sie müssen die Anbindung an die elektronische Patientenakte realisieren und dabei die vorgegebenen Schnittstellenspezifikationen sowie die Anforderungen aus Anlage 2 berücksichtigen.
- Die Authentisierung soll mittels digitaler Identität möglich sein.
- Die Forderung nach einem Zertifikat des BSI für den Nachweis der Erfüllung der Anforderung an Datensicherheit UND Datenschutz ersetzt die Eigenerklärung über die Anlage 1. Das gilt auch für Bestands-DiGAs. Sie sollten entsprechende Ressourcen vorsehen und die technischen Richtlinien des Bundesamts für Sicherheit in der Informationstechnik (BSI) berücksichtigen.
- Die Änderungen an der DiGAV bzw. dem zugrundeliegenden SGB V erfolgen recht schnell. Richten Sie ein Monitoring ein (SGB V, DVPMG, DiGAV, DiGAVÄndV), um auf dem Laufenden zu bleiben und die Fristen einzuhalten (s. u.).
- Redaktionelle Änderungen im DiGA-Verzeichnis sind keine wesentliche Änderung und können kostenfrei angezeigt werden.
1. Ergänzungen im Antrag
Im Antrag (s. DiGAV § 2 Antragsinhalt) müssen Informationen ergänzt werden zu
- den von der digitalen Gesundheitsanwendung verarbeiteten Daten,
- deren Darstellbarkeit mittels internationaler Semantikstandards und,
- bei Antragstellung ab 1. August 2022, zu deren Abbildbarkeit mittels der jeweils geltenden Festlegung für die semantische und syntaktische Interoperabilität von Daten der elektronischen Patientenakte,
- den menschenlesbaren Exportformaten aus der DiGA.
Umsetzungshinweise:
Die Hersteller müssen diese Punkte bei der Antragsstellung mit aufführen. Falls die Informationen nicht in deutscher Sprache vorliegen, müssen diese ins Deutsche übersetzt werden (Antragsinhalt, insb. die Inhalte, die in dem elektronischen Verzeichnis erscheinen.
Zertifizierung jetzt auch für Datenschutz und ISMS-Zertifizierung auch für Bestands-DiGAs
Ergänzung zu § „4 Anforderungen an Datenschutz und Datensicherheit“ bzw. § 7 „Nachweis durch Zertifikate“: Ab dem 1. April 2023 müssen digitale Gesundheitsanwendungen den Nachweis der Erfüllung der Anforderungen an den Datenschutz durch Vorlage eines Prüfzertifikats (z. B. vom BSI) erbringen. Das heißt, dass die Eigenerklärung über die Anlage 1 nicht mehr ausreichen wird. In § 7 wird klargestellt, dass die Verpflichtung, ab dem 1. April 2022 ein zertifiziertes Informationsmanagementsystem implementiert zu haben, auch für bereits gelistete DiGA-Hersteller gilt.
Umsetzungshinweise:
Die technische Richtlinie des BSI TR-03161 enthält einen expliziten Anforderungskatalog an umzusetzende Maßnahmen. Dieser wird vermutlich auch die Grundlage für die Prüfkriterien bilden. Für die Datensicherheit wurde bereits mit der letzten Änderung ein Prüfzertifikat vom BSI gefordert; jetzt gilt das für Datensicherheit UND Datenschutz. Es gibt also ein Zertifikat für das ISMS (z. B. ISO 27001), ein weiteres für den Nachweis, dass die Anforderungen an die Datensicherheit in der DiGA umgesetzt wurden, und ein drittes, wenn die Anforderungen an den Datenschutz in der DiGA umgesetzt wurden. Planen Sie das auch für Bestands-DiGA entsprechend ein.
Anforderungen an die Interoperabilität
Ergänzung zu § 6 „Interoperabilität“: Im SGB V wurde der Paragraph 355 ergänzt, welcher Festlegungen für die semantische und syntaktische Interoperabilität von Daten für die elektronische Patientenakte fordert. Bis zum 30. Juni 2022 soll die Kassenärztliche Bundesvereinigung entsprechende Festlegungen treffen. Bis dahin können auch offene, international anerkannte Schnittstellen und Semantikstandards verwendet werden.
Elektronische Patientenakte (ePA)
Ergänzung zu § 6a „Interoperabilität von digitalen Gesundheitsanwendungen mit der elektronischen Patientenakte“: Ab dem 1. Januar 2023 müssen DiGAs die verarbeiteten Daten – unter Einwilligung des Versicherten – in die elektronische Patientenakte des Versicherten übermitteln können, d. h. über eine entsprechende Schnittstelle verfügen, die von der Gesellschaft für Telematik vorgegeben wird. Dabei muss die semantische und syntaktische Interoperabilität berücksichtigt werden. Die Hersteller haben 6 Monate Zeit, um die Festlegungen an die semantische und syntaktische Interoperabilität von Daten umzusetzen, sobald diese von der Kassenärztlichen Bundesvereinigung veröffentlicht wurden.
Umsetzungshinweise:
Im Rahmen der Aktivitäten zur Überwachung nach dem Inverkehrbringen (PMS) sollte ein enges Monitoring der Änderungen der Festlegungen bzgl. der Schnittstellen sowie insgesamt der DiGA-bezogenen gesetzlichen Vorgaben vorgesehen werden. Empfehlenswert sind automatische Benachrichtigungen über Änderungen. Auch müssen noch weitere Spezifikationen geschrieben werden (z. B. für die sichere Identität).
Ab dem 1. Januar 2023 muss die elektronische Patientenakte (ePA) vom Hersteller unterstützt werden. Bis 1. Januar 2022 trifft die Gesellschaft für Telematik Festlegungen zur Schnittstelle.
Konkretisierung wesentlicher Änderungen
Änderungsanzeige: Es wird konkretisiert, was keine wesentlichen Veränderungen sind, nämlich „im Umfang geringfügige und lediglich redaktionelle Änderungen der Angaben und Informationen“ im DiGA-Verzeichnis. Diese Änderungen können dem BfArM durch einfache Anzeige mitgeteilt werden. Zuvor galten alle „im Verzeichnis für digitale Gesundheitsanwendungen bekannt gemachten Angaben und Informationen“ als wesentliche Änderungen. Weiterhin wird ergänzt, dass diese „einfache Anzeige“ von der Gebührenordnung ausgenommen ist.
Redaktionelle Änderungen
- Die in der Vergangenheit liegenden Fristangaben wurden entfernt (z. B. „spätestens ab dem 1. Januar 2021“).
- Es wird konkretisiert, dass bei „Interoperabilität“ von syntaktischer und semantischer Interoperabilität die Rede ist.
- Das Verzeichnis nach § 291e des SGB V wird jetzt als Interoperabilitätsverzeichnis nach § 385 des SGB V geführt (§ 291e des SGB V wurde aufgehoben)
- Der Verweis auf SGB V § 291b Absatz 1 Satz 7 wird ersetzt durch SGB V § 355 Absatz 2a in welchem jetzt festgelegt wird, dass die Kassenärztliche Bundesvereinigung bis zum 30. Juni 2022 die notwendigen Festlegungen für die semantische und syntaktische Interoperabilität von Daten aus DiGAs trifft, die in die elektronische Patientenakte übermittelt werden.
Änderungen in den Anlagen
- Anlage 1, Datenschutz, Punkt 13. Die Frage ob lokal auf dem genutzten IT-System abgelegte Daten und angelegte Dateien nach Beendigung der Nutzungs-Session der DiGA sicher gelöscht werden, auch wenn die nutzende Person die Nutzungs-Session nicht explizit beendet, entfällt. Es muss dafür also kein Nachweis erbracht werden
- Anlage 1, Datenschutz, Punkt 34. Es wird konkretisiert, dass es um „bei dem Hersteller gespeicherte personenbezogene Daten“ handelt, bei der Frage ob der Hersteller Maßnahmen zur Nachvollziehbarkeit von Änderungen an diesen Daten implementiert hat.
- Anlage 1, Datensicherheit, Punkt 1. Das ISMS muss erst ab 1 April 2022 auf Verlangen des BfArM, (statt bisher 1. Januar 2022) per Zertifikat nachgewiesen werden.
- Anlage 1, Datensicherheit, Punkt 15a kommt neu hinzu. Bis zum 01.01.2023 muss die DiGA die sichere digitale Identität nach § 291 Absatz 8 des SGB V unterstützen. Dafür wurde Punkt 6 für DiGAs mit besonders hohem Schutzbedarf gestrichen, die eine Authentisierung mittels elektronischer Gesundheitskarte und kontaktloser Schnittstelle forderte.
- Anlage 2, Interoperabilität Punkt 4a kommt neu hinzu mit der Frage ob eigene Profilierungen veröffentlicht sind
- Anlage 2, Interoperabilität Punkt 5 kommt neu hinzu. Die DiGA soll es ermöglichen (mit Einwilligung des Versicherten) die Daten in die ePA zu überführen und zwar auch automatisiert. Dieser automatisierte Mechanismus soll konfigurierbar sein und enden sobald die Verordnungsdauer beendet ist.
Danke an die Fachanwältin Sonia Seubert.
Das Johner Institut unterstützt Hersteller bei der Konzeption und Durchführung wissenschaftlicher Studien, die alle Anforderungen der DiGAV erfüllen. Nehmen Sie gleich Kontakt auf.
Änderungshistorie
- 2023-04-19: Hinweis ergänzt, dass das Vesta-Verzeichnis in den „Interoperabilitätsnavigator INA“ überführt wurde
- 2023-01-27: Artikel aktualisiert, Redundanzen mit anderen Artikel entfernt und Verweise ergänzt
- 2022-10-14: Unter 6. BfArM-Prüfkriterien für den Datenschutz verlinkt
- 2021-02-01: Änderungen durch BfArM-FAQ in Kapitel 6 ergänzt
- 2021-02-09: Link auf DiGAV statt auf Referentenentwurf
- 2021-07-05: Hinweis auf DVPMG eingefügt, Referenzen aktualisiert, Hinweise zu US-Cloud-Anbietern ergänzt
- 2021-11-19: Verordnung zur Änderung der Digitale Gesundheitsanwendungen-Verordnung (DiGAVÄndV) ergänzt (Aktuelles)



Lieber Herr Dr. Johner,
sehr gern mache ich von Ihrem Angebot Gebrauch, Ihnen eine Frage zu stellen. Und ich möchte Ihnen danken, dass Sie uns mit Ihrem Newsletter mit wichtigen Informationen versorgen.
Aussage:
BSI TR-03161 Sicherheitsanforderungen an digitale Gesundheitsanwendungen
Gegenstand der Technischen Richtlinie:
…
Diese Technische Richtlinie wendet sich an Hersteller von digitalen Gesundheitsanwendungen für mobile Endgeräte. …
Frage: immer wieder lese ich in Verordnungen, dass DiGa’s mit digigitalen Endgeräten verbunden werden und explizit auch so benannt werden. Das sind überwiegende Handys, manchmal auch Tablets.
Wunsere DiGa ist aber eine Desktop-Anwendung (Browser-Software), funktioniert zwar auch auf dem Handy, da es aber erst ab 10“ Bildschirmgröße Sinn macht, sind Handy’s ungeeignet.
Jetzt die genaue Frage:
Wie umfassend oder ausschließend meint der Gesetzgeber hier „für mobile Endgeräte“? Sind Desktop-Computer nicht dabei? Werden Online-Anwendungen anders behandelt als Apps aus dem App-Shop?
Danke vorab und mit besten Wünschen aus Hamburg
Hannes Meeves
040 72104688
Sehr geehrter Herr Meeves,
danke für die spannende Frage!
DiGAs umfassen definitiv auch „Nicht-Apps“ wie beispielsweise browserbasierte Anwenndungen.
Dass die TR-03161 nur DiGAs für mobile Endgeräte adressiert, ändert daran nichts. Es ist nicht einmal gesagt, dass damit überhaupt DiGAs per definitionem des DVGs gemeint sind. Beispielsweise wäre für eine Klasse-III-App dieser Leitfaden ebenso anzuwenden.
Danke für die Frage, die diese Klarstellung ermöglicht!
Viele Grüße, Christian Johner
Ein dickes Lob, Herr Johner !
Auch wenn ich nur bedingt mit Ihrer Zielgruppe zu tun habe, leisten mir Ihre vorzüglich recherchierten Newsletter immer wieder gute Dienste bei Interpretation und Bewertung von regulativen Sachverhalten im engeren oder weiteren Kontext der jeweiligen Gesetzgebung.
Mit freundlichem Gruss,
Stefan Glaus
Danke, lieber Herr Glaus, für Ihre Rückmeldung!
Ich freue mich riesig darüber! Das gibt Kraft und Motivation!
Nochmals vielen Dank!
Sehr geehrter Herr Johner,
die DiGAV löst mMn auch die Anwendung der IEC 82304 aus – wie sehen Sie das? Haben Sie ggf. schon analysiert, ob/wo sich die DiGAV und die IEC 82304 ergänzen (oder vielleicht widersprechen)?
Viele Grüße
Raimund Mödlhammer
Sehr geehrter Herr Mödlhammer,
die DiGAV löst im Wesentlichen drei Dinge aus:
Wenn, dann wäre es die MDR, die die IEC 82304 „auslöst“. Da es momentan aber keine harmonisierte Normen gibt, ist diese Frage umstritten. Die IEC 82304 ist allerdings im letzten Entwurf der Liste der zu harmonisierten Normen nicht mehr enthalten.
Ein Auditor kann somit nur mit allgemeiner Bezugnahme zum Stand der Technik Anforderungen der IEC 82304 einbeziehen. Aber eine Konformität mit der Norm kann er nicht verlangen.
Viele Grüße, Christian Johner
Unfreiwillige DiGA?
Hallo Herr Dr. Johner,
in einschlägigen App-Stores finden sich Andwendungen mit Aussagen wie z. B. „Mit der führenden Schnarch-Analyse-App können Sie Ihr Schnarchen aufzeichnen, messen und reduzieren. Von Ärzten empfohlen!“ -> [apnoes] Schnarchen reduzieren“ und „von Ärzten empfohlen“ = eine Therapie?!
Auf der Hersteller-Website findet sich dann der Disclaimer: „XXX is not a medical device, nor is it intended to replace professional medical advice and/or diagnosis. XXX is not intended to diagnose or treat sleep apnea or any illness/medical condition.“
Ich befürchte, dass das nicht ausreicht und hier dennoch eine [unfreiwillige] DiGA (Medizinprodukt!) unterstellt werden könnte. Das hätte dann erhebliche Konseqenzen für den Hersteller (und die Anwender).
Wie sehen Sie das?
Vielen Dank für Ihre Einschätzung und freundliche Grüße
Raimund Mödlhammer
Lieber Herr Mödlhammer,
ich verstehe Ihren Punkt: man könnte einen Widerspruch sehen zwischen dem Anspruch („Schnarchen reduzieren“) und der Aussage, dass es kein Medizinprodukt ist.
Da es aber mit der Verneinung, dass ein Medizinprodukt sei, eine ziemlich klare Aussage ist und von Schnarchen und eben nicht der Krankheit (Apnoe) gesprochen wird, halte ich das für unkritisch. Wenn wir auf diesem Niveau uns auf die Suche begeben, werden wir wahrscheinlich Hunderte größere Problemfälle finden.
Das es eine DIGA ist, würde ich gar nicht so sehen, da nich jedes digitale Medizinprodukt eine DIGA ist.
Beste Grüße, Christian Johner
Sehr geehrter Herr Dr. Johner,
vielen Dank für Ihre schnelle Antwort.
Das BfArM äußert sich recht streng zu diesem Punkt:
„Erklärungen wie z. B. ein Vermerk im App-Store ‚Dies ist kein Medizinprodukt‘ umgehen die o. g. Kriterien [für ein Medizinprodukt] NICHT […]“.
https://www.bfarm.de/DE/Medizinprodukte/Abgrenzung/MedicalApps/_node.html.
Das BfArM behält sich vor, bei deren Klassifizierung (Medizinprodukt ja/nein) eine „Medizinische Zweckbestimmung“ auch der Kennzeichnung und/oder dem Werbematerial zu entnehmen. Dabei wird auf „Anhaltsbegriffe“ bzw. „Anhaltsfunktionen“ geachtet: z. B. analysieren, messen, interpretieren, Entscheidungsunterstützung …
Es bleibt spannend, ob/wie das in der Praxis gelebt wird, falls sich eine Wellness/Lifestyl/Fittness-App hier (versehentlich) zu weit aus dem Fenster lehnt und damit unfreiwillig zum Medizinprodukt wird…
Hallo,
da DIGA Medizinprodukte sind: Sind die Krankenkassen Betreiber im Sinne der MPBetreibV? Haben die Krankenkassen einen Rahmenvertrag geschlossen, mit dem sie ihre Betreiberpflichten auf die Hersteller übertragen haben? Oder gibt es zu den Betreiberpflichten spezielle Regelungen in der DIGAV oder sonstwo?
Herzliche Grüße
Jörg Stemmler
Sehr geehrter Herr Stemmler,
danke für die sehr spannende Frage!
Auf Ihre Frage gibt es keine allgemein gültige Antwort. Denn die Kassen können verschiedene Rollen einnehmen. In den meisten Fällen sind sie keine Betreiber, sondern „nur“ die Kostenerstatter. Teilweise treten die Krankenkassen aber auch in anderen Rollen auf.
Für DIGAs gelten keine speziellen Betreiberpflichten aus der MPBetreibV. Denn die MPBetreibV macht keinen Unterschied. Die DIGAV unterscheidet nicht, ob ein Betreiber eine Kasse ist. Allerdings ergänzt die DIGAV Betreiberpflichten u.a. im Kontext der IT-Security.
Mit den besten Grüßen, Christian Johner