
Mit der Verordnung 2021/2282 hat die EU ein Instrument geschaffen, um die Bewertung von Gesundheitstechnologien (Health Technology Assessment, HTA) unionsübergreifend zu vereinheitlichen. Dies soll es den Mitgliedstaaten leichter machen, über die gesundheitspolitischen Konsequenzen neuer Gesundheitstechnologien und deren Preisgestaltung zu entscheiden. Für Hersteller bzw. Entwickler entfällt im Idealfall doppelter Aufwand. Doch der Unionsgesetzgeber ist mit der HTA-Verordnung auch einige Kompromisse eingegangen, die den Effekt mindern könnten.
In diesem Beitrag erfahren Sie, was die wichtigsten Inhalte der HTA-Verordnung sind.
Autorin: Dr. Anja Segschneider
1. HTA: Die Grundlagen
a) Definition
Die Bewertung von Gesundheitstechnologien (Health Technology Assessment — im Folgenden „HTA“) ist ein wissenschaftlicher evidenzbasierter Prozess, mit dessen Hilfe zuständige Behörden die relative Wirksamkeit neuer oder bestehender Gesundheitstechnologien bestimmen können.
Im Zentrum der HTA-Verordnung steht der Vergleich einer Gesundheitstechnologie (Arzneimittel und Medizinprodukte, In-Vitro-Diagnostika, medizinische Verfahren, Maßnahmen zur Prävention von Krankheiten sowie Diagnose- und Behandlungsverfahren) mit anderen Technologien. Hierdurch können beispielsweise die Wirksamkeit, Sicherheit sowie die ökonomischen, sozialen und ethischen Aspekte von Gesundheitstechnologien besser eingeschätzt werden. Entsprechend umfasst die HTA-Verordnung auch klinische (z. B. Wirksamkeit, Sicherheit) und nichtklinische Aspekte (z. B. ökonomische, ethische, organisatorische, soziale und rechtliche).
b) Nutzen für Entscheidungsträger
In der Praxis dienen HTAs vor allem als Grundlage für gesundheitspolitische Entscheidungen der Staaten, etwa für die Zuteilung von Haushaltsmitteln im Gesundheitsbereich. HTAs sind auch entscheidend dafür, welche Technologien zu welchen Konditionen beim Einsatz in der Gesundheitsversorgung erstattungsfähig sind. Die einschlägigen Regelungen hierfür finden sich in Deutschland v. a. im SGB V.
Darüber hinaus sind HTAs auch für andere Beteiligte des Gesundheitswesens relevant: Etwa für Kliniken und Gesundheitspersonal, die auf Grundlage einer HTA entscheiden können, welche Technologie in der Therapie zum Einsatz kommt. Indirekt können HTAs also die Gesundheit von Patientinnen und Patienten verbessern, indem sie zum Zugang zu neuen und/oder verbesserten Technologien beitragen.
c) Nutzen für Hersteller bzw. Entwickler
Für Hersteller sind die HTAs relevant, weil sie Entscheidungen über die Erstattungsfähigkeit ihrer Technologie beeinflussen. Die Erstattung wiederum wirkt sich darauf aus, wie stark Kliniken und Praxen die Technologie nachfragen.
Zudem profitieren die Hersteller bei der Erstellung ihrer klinischen Bewertungen. Denn die HTAs enthalten für die relevanten/äquivalenten Produkte eine
- sorgfältig zusammengestellte Literaturrecherche und Literaturübersicht,
- aufbereitete Beschreibung des State of the Art,
- Zusammenstellung von Nutzen und Risiken sowie
- Bewertung des Nutzen- und Risikoverhältnisses.
All diese Aspekte müssen die Hersteller als Entwickler der Technologie in der klinischen Bewertung berücksichtigen und können diese ganz oder teilweise aus den HTAs übernehmen. Das spart nicht nur Arbeit, sondern verschafft sowohl den Herstellern als auch den Benannten Stellen zusätzliche Gewissheit, dass die klinische Bewertung korrekt und vollständig ist.
2. Allgemeines zur Verordnung 2021/2282
a) Ziel
HTA-Verfahren finden in der EU bisher auf nationaler Ebene statt. Dies kann jedoch dazu führen, dass verschiedene Mitgliedstaaten gleichzeitig die Entwickler von Gesundheitstechnologien auffordern, unterschiedliche Daten einzureichen. Dies bedeutet also mehrfachen Aufwand für Hersteller, die mitunter Unterlagen doppelt einreichen müssen. Außerdem ist der Ausgang der nationalen HTA-Verordnungen u. U. unterschiedlich. In einem Land kann eine Technologie z. B. erstattet werden, im Nachbarland dagegen nicht.
Daher bietet die neue HTA-Verordnung 2021/2282 “über die Bewertung von Gesundheitstechnologien und zur Änderung der Richtlinie 2011/24/EU” eine Harmonisierung auf EU-Ebene. Sie verspricht den Unternehmen eine Reduzierung des Aufwands, indem ein Großteil der Unterlagen nur einmal auf EU-Ebene statt mehrfach in verschiedenen Staaten eingereicht werden sollen.
Ziel ist es auch, die methodische Vorgehensweise bei HTAs zu vereinheitlichen. Mit gemeinsamen klinischen Bewertungen von Gesundheitstechnologien wie Pharmazeutika, Medizinprodukten und In-Vitro-Diagnostika soll für die Mitgliedstaaten eine unionsübergreifende, vergleichbare Entscheidungsgrundlage u. a. für die Erstattungsfähigkeit geboten werden.
Ziele der HTA-Verordnung sind:
- Einheitliche Maßstäbe für das HTA-Verfahren
- EU-finanzierte Basis für gemeinsame Verfahren
- Zentrale Koordinierung
- Aufbau einer Plattform für Zusammenarbeit
Mit der Verordnung soll außerdem ein hohes Maß an Gesundheitsschutz für Patienten und Anwender erreicht und dabei das reibungslose Funktionieren des Binnenmarkts in Bezug auf Arzneimittel, Medizinprodukte und In-vitro-Diagnostika gewährleistet werden.
b) Anwendungsbereich
Die HTA-Verordnung setzt einen Fokus auf die klinischen Aspekte der HTAs, d. h. auf die relative klinische Wirksamkeit und die relative klinische Sicherheit einer neuen Gesundheitstechnologie gegenüber bestehenden Technologien. Diese EU-HTA-Verordnung soll sich nicht nur auf den klinischen Teil der HTAs beschränken, sondern auch absolut objektiv sein. Die Einordnung und Bewertung der auf EU-Ebene erzielten Erkenntnisse bleibt den Mitgliedstaaten überlassen.
Im Detail haben die Hersteller bzw. Entwickler von Gesundheitstechnologien also in Zukunft mit zwei HTA-Systemen zu tun: dem der EU und einem nationalen.
- EU-HTA: Liefert einen Bericht über objektive klinische Aspekte der HTA
- Nationale HTA: Nutzt die EU-HTA als Grundlage und bezieht zusätzliche Aspekte ein (etwa Ökonomie und Ethik). Die Bewertung beider Teile der HTAs bleibt nationalen Stellen überlassen.
Die HTA-Stellen der EU werden außerdem gemeinsame wissenschaftliche Konsultationen durchführen, um Technologieentwickler im Hinblick auf die Konzeption klinischer Studien zu beraten, damit sie geeignete Nachweise erbringen.
Vielversprechende Gesundheitstechnologien sollen durch strategische Früherkennung („Horizon Scanning“) frühzeitig ermittelt werden. Darüber hinaus soll die freiwillige Zusammenarbeit auf Ebene der Mitgliedstaaten gefördert werden.
Im Detail bietet die neue HTA-VO:
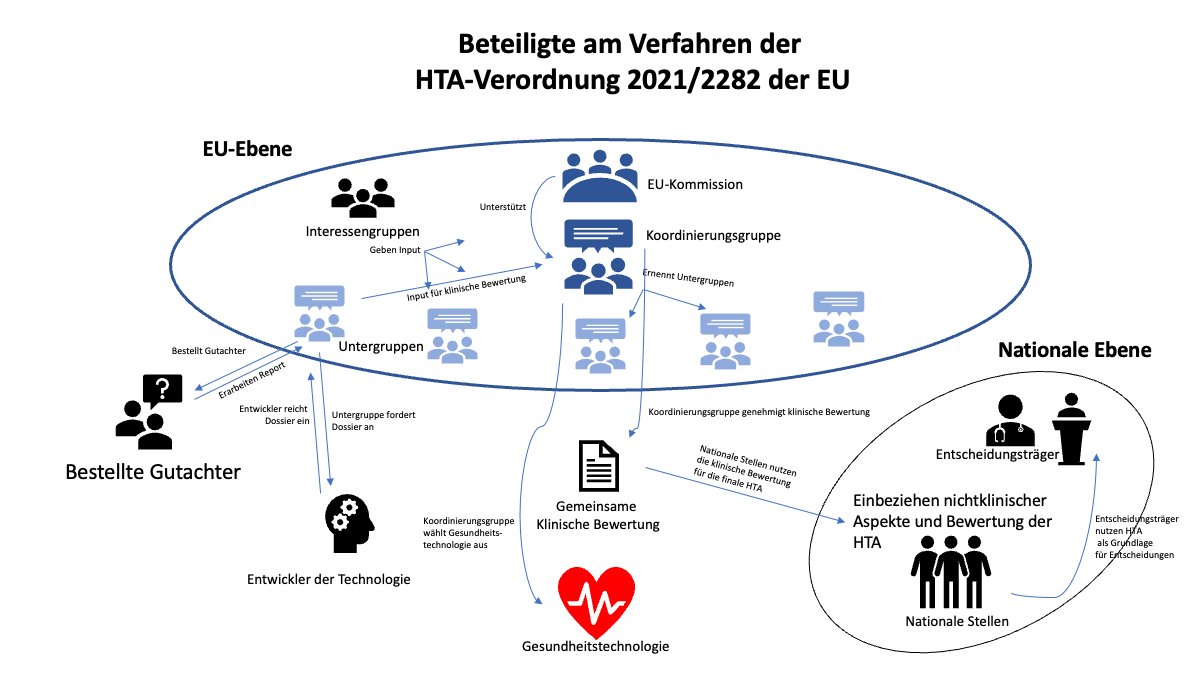
- Koordinierungsgruppe (Art. 3 bis 6)
Kernstück der HTA-Verordnung ist die Einrichtung einer Koordinierungsgruppe, die die gesamte Koordination der HTAs auf europäischer Ebene und der Zusammenarbeit von Staaten, Unternehmen und Verbänden koordinieren soll.
- Gemeinsame klinische Bewertung (Art. 7 bis 15)
Die Koordinierungsgruppe bzw. deren Untergruppen bestellen Gutachter, die gemeinsame klinische Bewertungen von Gesundheitstechnologien vornehmen. Hersteller, also Entwickler der Technologie, müssen dazu ein Dossier einreichen. Diese können z. B. den EU-Mitgliedstaaten als Entscheidungsgrundlage für ihre Gesundheitspolitik dienen.
- Gemeinsame wissenschaftliche Beratung (Art. 16 bis 21)
Gemeinsame wissenschaftliche Beratungen können Entwickler von Gesundheitstechnologien, für die noch keine gemeinsame klinische Bewertung geplant ist, beantragen. Bei der wissenschaftlichen Beratung wird eine Evidenzbasis geschaffen, die als Grundlage für eine spätere gemeinsame klinische Bewertung dient.
- Neu entstehende Gesundheitstechnologien (Art. 22)
Die Koordinierungsgruppe soll neu entstehende Gesundheitstechnologien beobachten und entscheiden, welche für eine gemeinsame klinische Bewertung herangezogen werden.
- Freiwillige Zusammenarbeit (Art. 23)
Die Kommission will auch über die gemeinsamen klinischen Bewertungen und wissenschaftlichen Beratungen hinaus die Zusammenarbeit zwischen den Mitgliedstaaten in Bezug auf neue Gesundheitstechnologien fördern.
- Netzwerk der Interessenträger (Art. 29)
Es soll ein Netzwerk von Interessenträgern eingerichtet werden, die Input für das Verfahren geben können. Hierfür können sich Interessensvereinigungen nach offizieller Aufforderung bewerben.
- IT-Plattform (Art. 30)
Der gesamte Informationsaustausch soll über eine neu geschaffene IT-Plattform erfolgen.
c) Fristen
Die neue HTA-Verordnung 2021/2282 ist im Januar 2022 in Kraft getreten. Sie ist ab dem 12. Januar 2025 gültig und wird bis 2028 schrittweise ausgebaut.
“Im Interesse einer reibungslosen Ein- und Durchführung der gemeinsamen klinischen Bewertungen auf Unionsebene sowie der Qualität dieser Bewertungen sollte zunächst eine geringe Anzahl von Arzneimitteln einer gemeinsamen klinischen Bewertung unterzogen werden. Drei Jahre nach Geltungsbeginn dieser Verordnung sollte die Zahl der gemeinsamen klinischen Bewertungen schrittweise erhöht werden.”
HTA-VO Erwägungsgrund 53
Vorrangig werden Arzneimittel zur Behandlung von Krebs berücksichtigt. Ab 2025 sollen auch bereits Medizinprodukte und IVD “mindestens alle zwei Jahre” zur Beurteilung herangezogen werden; angesichts des Vorzugs von Arzneimitteln werden dann vermutlich noch keine oder nur sehr wenige Entwickler neuer Technologien in den Bereichen Medizintechnik und IVD betroffen sein.
Mit dem Aufschub des Geltungsbeginns von drei Jahren soll sichergestellt werden, dass genügend Zeit für den Aufbau des organisatorischen Rahmens der HTA-Verordnung (z. B. der Koordinierungsgruppe und ihrer Untergruppen, des Netzwerks der Interessenträger) verbleibt. Zudem basieren weite Teile des EU-HTA-Prozesses auf delegierten Rechtsakten und Leitlinien, die die zuständigen Stellen erst noch erlassen müssen.
Die erste Sitzung der Koordinierungsgruppe ist für Juni 2022 geplant.
d) Hintergrund
Bereits vor Erlass der Verordnung gab es Ansätze, die HTAs auf europäischer Ebene über das EU-kofinanzierte European Network for Health Technology Assessment (EUnetHTA) zu koordinieren. Dies erfolgte jedoch auf freiwilliger Basis und war wenig effektiv. Da die EU-Verordnung in den Mitgliedstaaten unmittelbar bindend ist, soll sie nun eine höhere Wirksamkeit entfalten.
3. Inhalte der Verordnung (EU) 2021/2282
a) Koordinierungsgruppe (Art. 3 bis 6)
Eines der zentralen Elemente der HTA-Verordnung ist die Einrichtung einer Koordinierungsgruppe. Diese steht im engen Austausch mit der EU-Kommission. Zwar gilt die HTA-Verordnung erst ab 2025, doch sollen erste Sitzungen der Koordinierungsgruppe bereits im Juni 2022 stattfinden.
Die Koordinierungsgruppe soll folgendermaßen ausgestaltet sein:
- Die EU-Mitgliedstaaten benennen die Mitglieder
- Jeder Staat hat eine Stimme
- Die Mitglieder der Koordinierungsgruppe ernennen wiederum nationale oder regionale Untergruppen, in denen HTA-Expert:innen vertreten sind.
Aufgaben
Zu den Aufgaben der Koordinierungsgruppe zählt vor allem
- Koordinierung der Untergruppen
- Ermittlung neu entstehender Gesundheitstechnologien
- Erstellung strategischer und methodischer Leitfäden für die nationalen Untergruppen
- Vorgabe von Verfahrensschritten und Fristen für die Durchführung und Aktualisierungen gemeinsamer klinischer Bewertungen und wissenschaftlicher Beratungen
- Einbeziehen von Interessenverbänden
b) Gemeinsame klinische Bewertung (Art. 7 bis 15)
Ein weiterer zentraler Bestandteil der HTA-Verordnung sind gemeinsame klinische Bewertungen (Joint Clinical Assessment, JCA). Diese beschränken sich jedoch auf die Beschreibung der Gesundheitstechnologie sowie die rein sachliche Prüfung ihrer technischen und klinischen Eigenschaften. Die Bewertung aller nichtklinischen Aspekte und die Schlussfolgerungen daraus, also etwa die Erstattungsfähigkeit, bleibt weiter den Mitgliedstaaten überlassen.
Anwendungsbereich
Nach Art. 7 der HTA-Verordnung wählt die Kommission ab 2025 mindestens alle zwei Jahre die Medizinprodukte und In-Vitro-Diagnostika für eine gemeinsame klinische Bewertung aus. Dies gilt allerdings nur für
- Medizinprodukte der Klassen IIb oder III
gemäß Artikel 51 MDR (Verordnung (EU) 2017/745), die die zuständigen Expertengremien im Rahmen des Konsultationsverfahrens im Zusammenhang mit der klinischen Bewertung gemäß Artikel 54 MDR ein wissenschaftliches Gutachten abgegeben haben.
- In-vitro-Diagnostika der Klasse D
gemäß Artikel 47 IVDR (Verordnung (EU) 2017/746), für die die zuständigen Expertengremien im Rahmen des Verfahrens gemäß Artikel 48 Absatz 6 der genannten Verordnung ihre Standpunkte vorgelegt haben.
Kriterien für die Auswahl von Medizinprodukten und IVD für eine EU-HTA sind dabei:
- Ungedeckter medizinischer Bedarf
- Neuigkeitswert (erstes Produkt einer neuen Produktkategorie)
- Mögliche Auswirkungen auf Patienten, öffentliche Gesundheit oder Gesundheitssysteme
- Einbeziehung von künstlicher Intelligenz, Technologien des maschinellen Lernens oder Algorithmen
- Relevanz der Technologie im internationalen Raum
- Großer, unionsweiter Mehrwert (etwa Steigerung der Gesundheit der Bevölkerung, wirtschaftlicher Mehrwert etc.)
Ablauf
Die gemeinsame klinische Bewertung verläuft in den folgenden Schritten:
- Einleitung durch die Untergruppe (Art. 8)
Die Untergruppen leiten die gemeinsame klinische Bewertung ein, indem sie einen geeigneten Fachgutachter und einen Mitgutachter bestellen, die aus unterschiedlichen Mitgliedstaaten stammen.
- Die Untergruppe legt den Bewertungsumfang fest (Art. 8)
Der Bewertungsumfang schließt insbesondere alle relevanten Parameter für die Bewertung ein in Bezug auf:- Patientenpopulation
- Intervention beziehungsweise Interventionen
- Komparator beziehungsweise Komparatoren
- Gesundheitsbezogene Endpunkte
- Vom Entwickler der Gesundheitstechnologie bereitgestellte Informationen
- Einbringungen von Patienten, klinischen Experten und anderen einschlägigen Sachverständigen
- Belange der Mitgliedstaaten
- Hersteller bzw. Entwickler der Gesundheitstechnologie legt ein Dossier vor (Art. 10, 11)
Die Kommission fordert den Entwickler auf, ein Dossier mit allen nötigen Informationen zur Technologie zusammenzustellen und einzureichen. Der Inhalt wird von Anhang II der HTA-Verordnung und Delegierte Akte näher bestimmt. Ein Unternehmen darf dabei keine Nachweise auf nationaler Ebene einreichen, die bereits auf Unionsebene vorliegen.
Wenn im Laufe des Bewertungsverfahrens neue klinische Daten verfügbar werden, hat der betreffende Entwickler der Gesundheitstechnologie die Koordinierungsgruppe darüber proaktiv zu informieren (Art. 11).
- Vornahme der Bewertung
Gutachter und Co-Gutachter bewerten die Technologie auf Grundlage des Dossiers und des festgelegten Bewertungsumfangs.
Gutachter und der Mitgutachter können auch auf Datenbanken und andere Quellen klinischer Informationen, Daten, Analysen oder sonstige Nachweise (z. B. Patientenregister) zurückgreifen. - Erstellen des Berichts
Gutachter und Mitgutachter erstellen einen analytischen Bewertungsbericht sowie einen zusammenfassenden Bericht (JCA Report; Art. 11 Abs. 1).
Nach Artikel 11 werden die Berichtentwürfe für Medizinprodukte und IVD von der Koordinierungsgruppe im Einklang mit den Fristen gemäß Artikel 3 Absatz 7 Buchstabe e und Artikel 15 Absatz 1 Buchstabe b gebilligt. Diese besagen letztlich, dass die Koordinierungsgruppe die Fristen selbst festlegt.
- Anmerkungen der Mitglieder der Untergruppe, des Herstellers und anderer werden eingearbeitet
Der überarbeitete Entwurf wird an die Koordinierungsgruppe übermittelt. - Finale Überarbeitung durch Koordinierungsgruppe (Art. 12)
- Bericht wird auf dafür eingerichteter IT-Plattform von der Kommission veröffentlicht.
c) Gemeinsame wissenschaftliche Beratung (Art. 16 bis 21)
Die gemeinsamen wissenschaftlichen Beratungen sind den gemeinsamen klinischen Bewertungen vorgeschaltet. Für gemeinsame wissenschaftliche Beratungen kommen Gesundheitstechnologien in Frage, für die zwar noch keine gemeinsame klinische Bewertung geplant ist, die aber voraussichtlich Gegenstand gemeinsamer klinischer Bewertungen sein werden.
Im Unterschied zur gemeinsamen klinischen Beratung findet die gemeinsame wissenschaftliche Beratung also statt,
- wenn die Technologie noch nicht von der Koordinierungsgruppe ausgewählt wurde,
- sie aber vielversprechend ist, sowie
- auf Antrag des Herstellers bzw. Entwicklers.
Die wissenschaftliche Beratung wird also vor allem für Entwickler interessant sein, die ihre Technologie in den Fokus der Koordinierungsgruppe rücken möchten.
Gemeinsame wissenschaftliche Beratung auf Antrag
Entwickler von Gesundheitstechnologien können eine gemeinsame wissenschaftliche Beratung beantragen. Eine Zu- oder Absage auf den Antrag des Entwicklers bzw. Herstellers soll innerhalb von 15 Arbeitstagen nach Ende eines Antragszeitraums erfolgen.
Kriterien für die Auswahl umfassen:
- Ungedeckter medizinischer Bedarf
- Erstes Produkt einer neuen Produktkategorie
- Mögliche Auswirkungen auf Patienten, öffentliche Gesundheit oder Gesundheitssysteme
- Signifikante grenzüberschreitende Dimension
- Großer, unionsweiter Mehrwert
- Prioritäten der Union in der klinischen Forschung
Nutzen der gemeinsamen wissenschaftlichen Beratung
Nach Art. 16 der HTA-Verordnung führt die Koordinierungsgruppe gemeinsame wissenschaftliche Beratungen durch, um mit Entwicklern Informationen über deren Pläne für bestimmte Gesundheitstechnologien auszutauschen. Diese Beratungen sollen die Evidenzgenerierung für eine anschließende gemeinsame klinische Bewertung erleichtern.
Die gemeinsame wissenschaftliche Beratung umfasst ein Treffen mit dem Entwickler und endet mit einem Abschlussdokument, in dem die abgegebene wissenschaftliche Empfehlung erläutert wird.
d) Neu entstehende Gesundheitstechnologien (Art. 22)
Die Koordinierungsgruppe verfolgt die Entwicklung neuer Technologien jedoch nicht nur auf Antrag. Über neu entstehende Gesundheitstechnologien sollen regelmäßig Berichte verfasst werden. Diese stützt sich auf bestehende wissenschaftliche Berichte oder Initiativen bezüglich neu entstehender Gesundheitstechnologien sowie auf Informationen aus einschlägigen Quellen (Register für klinische Studien und wissenschaftliche Berichte, Angaben der Europäischen Arzneimittelbehörde EMA, die nun auch eine Rolle bei Medizinprodukten übernimmt etc.).
e) Freiwillige Zusammenarbeit (Art. 23)
Die Kommission will auch über die gemeinsamen klinischen Bewertungen und wissenschaftlichen Beratungen hinaus die Zusammenarbeit zwischen den Mitgliedstaaten in Bezug auf neue Gesundheitstechnologien und den Austausch von wissenschaftlichen Informationen fördern. Auch diesen Austausch soll die Koordinierungsgruppe voranbringen.
f) Netzwerk der Interessenträger (Art. 29)
Die Kommission richtet ein Netzwerk der Interessenträger ein. Das Netzwerk der Interessenträger soll die Koordinierungsgruppe und ihre Untergruppen auf Anfrage bei ihrer Arbeit unterstützen.
Wie das Netzwerk entstehen soll
Es wird eine offene Aufforderung an Interessenverbände geben, sich für das Netzwerk zu bewerben. Diese wird sich insbesondere richten an:
- Patientenvereinigungen, Verbraucher- und Nichtregierungsorganisationen aus dem Gesundheitsbereich
- Entwickler von Gesundheitstechnologien
- Angehörige von Gesundheitsberufen
Die Auswahlkriterien werden im Rahmen der offenen Aufforderung zur Einreichung von Bewerbungen festgelegt und beinhalten Folgendes:
- Nachweise für die laufende oder geplante Beteiligung an der Entwicklung von HTA
- Für das Netzwerk der Interessenträger relevante Fachkompetenz
- Geografische Abdeckung mehrerer Mitgliedstaaten
- Kommunikations- und Verbreitungskapazitäten
g) IT-Plattform (Art. 30)
Mit der HTA-Verordnung plant die Kommission eine weitere IT-Plattform, über die die Kommunikation ablaufen soll. Diese Plattform soll nach Art. 30 aus zwei Kernstücken bestehen:
- Einer Website, auf der die Berichte und weitere Informationen veröffentlicht werden
- Eine Plattform, über die datensicher Informationen ausgetauscht werden können. Diese Plattform soll etwa für den Austausch von Daten zwischen der Koordinierungsgruppe und den Untergruppen, zwischen den Mitgliedstaaten und der Koordinierungsgruppe oder zum Datenaustausch mit Entwicklern genutzt werden.
4. Auswirkung der HTA-VO auf Hersteller
a) Was sind die Pflichten?
In erster Linie müssen Hersteller, die eine neue Gesundheitstechnologie entwickelt haben, auf Anfrage der EU Unterlagen zu ihrer Technologie bereitstellen. Welche Unterlagen im Einzelnen angefordert werden, ist noch nicht festgelegt.
Diese Aufforderung der Koordinierungsstelle zum Einreichen von Unterlagen enthält eine Frist sowie genauere Angaben über Form und Umfang der einzureichenden Unterlagen.
b) Was passiert, wenn Entwickler die Pflicht nicht erfüllen?
Kommt ein Entwickler der Aufforderung nicht oder nicht vollständig nach, erhält er zunächst eine zweite Aufforderung. Lässt der Entwickler auch diese Frist verstreichen, stellt die Koordinierungsgruppe die HTA für die Technologie ein und veröffentlicht eine entsprechende öffentliche Bekanntmachung auf der IT-Plattform.
Allerdings kann die Koordinierungsgruppe die HTA erneut einleiten, wenn der Entwickler entsprechende Unterlagen auf nationaler Ebene eingereicht hat.
c) Welchen Nutzen verspricht die neue EU-HTA-Verordnung?
In erster Linie vereinheitlicht die HTA-Verordnung die Maßstäbe, mit denen europaweit HTAs durchgeführt werden. Damit nutzt die Verordnung potenziell vor allem Patientinnen und Patienten, die sich auf ein einheitlicheres Niveau der Beurteilungen in allen Mitgliedstaaten verlassen können. Auch den Entscheidungsträgern aus Politik und Gesundheitswesen könnten diese Vereinheitlichung und die damit entwickelten transparenten Maßstäbe und vergleichbaren Bewertungen in den einzelnen Mitgliedstaaten die Arbeit erleichtern.
Vorteile für Hersteller
Die VO verspricht in ihren Erwägungsgründen auch den Herstellern Erleichterungen, insbesondere kleineren und mittelständischen Unternehmen. Die Unternehmen sollen nicht durch mehrere Staaten zur Einreichung von unterschiedlichsten HTA-Daten mit unterschiedlichsten Fristen aufgefordert werden können. Dass dies explizit in den Erwägungsgründen genannt wird, hat wohl auch damit zu tun, dass die Verordnung im Vorfeld u. a. dafür kritisiert wurde, dass sie (zusätzlich zur MDR) erneut einen Mehraufwand für die Hersteller bedeuten könnte.
d) Werden die Versprechen gehalten?
Die HTA-Verordnung nennt in der Einleitung als Folgen geringeren Aufwand und einheitliche Beurteilungen. Ob diese Versprechen am Ende gehalten werden, lässt sich durchaus in Frage stellen. Denn letztlich bleibt ein großer Teil der Entscheidungen den Mitgliedstaaten selbst überlassen. Mehr noch, die Verordnung stellt explizit klar, dass auf nationaler Ebene zusätzliche Verfahren durchgeführt werden dürfen, für die die Staaten auch zusätzliche Daten von Entwicklern anfordern dürfen.
“Sollten für ergänzende klinische Analysen zusätzliche Informationen, Daten, Analysen und sonstige Nachweise erforderlich sein, so sollte der Mitgliedstaat den Entwickler der Gesundheitstechnologie auffordern können, die erforderlichen Informationen, Daten, Analysen und sonstigen Nachweise vorzulegen.”
HTA-VO Erwägungsgrund 15
Es ist auch fraglich, wie sehr sich der HTA-Prozess insgesamt verzögern wird, wenn der erste Schritt auf EU-Ebene und der zweite auf nationaler Ebene erfolgt. BVMed ging in einer Stellungnahme aus 2018 noch von zwölf Monaten Verzögerung aus.
Ob die HTA-Verordnung am Ende also mehr oder weniger Aufwand für Unternehmen bedeutet, wird sich mit der Zeit zeigen.
e) Müssen sich Hersteller sorgen?
Kurz gesagt: Erst einmal nicht. Mit den HTAs beginnen die EU-Stellen erst im Januar 2025. Vorrangig sollen Arzneimittel zur Behandlung von Krebs der EU-HTA unterzogen werden. Außerdem sollen zunächst nur wenige HTAs durchgeführt werden. Es liegt also nahe, davon auszugehen, dass Entwickler aus den Bereichen Medizintechnik und IVD zunächst nur wenig betroffen sind. Allerdings sollten Hersteller von Medizinprodukten die Entwicklungen in den kommenden Jahren unbedingt im Auge behalten.
5. Zusammenfassung und Fazit
Die HTA-Verordnung 2021/2282 versucht sich an einer Vereinheitlichung des HTA-Prozesses auf EU-Ebene, geht dabei aber viele Kompromisse ein. Die Verordnung macht an mehreren Stellen klar, dass die finale Entscheidung über die Gesundheitstechnologien weiterhin den Mitgliedstaaten überlassen bleibt. Dieser Kompromiss war wegen der in den EU-Verträgen festgelegten Hoheit der Mitgliedstaaten über das Gesundheitswesen nötig und schwächt die Wirksamkeit der HTA-Verordnung letztlich ab. Die EU-HTA beschränken sich auf die klinische Dimension und rein sachliche Analysen. Die Beurteilung des Zusatznutzens und das Ziehen von Konsequenzen aus der Beurteilung (etwa Erstattungsfähigkeit) erfolgt weiter auf nationaler Ebene.
Das bedeutet jedoch nicht, dass die EU-HTA nach Verordnung 2021/2282 vollkommen irrelevant sind.
Vor allem werden die Berichte der Koordinierungsgruppe als Grundlage für Entscheidungen der Mitgliedstaaten dienen. Diese müssen die EU-Berichte im Rahmen der nationalen HTA in angemessener Weise berücksichtigen (Art. 13 Abs. 1). Dies wird sich vor allem bei der Entscheidung über die Nutzung und die Kostenübernahme für eine Gesundheitstechnologie im deutschen Gesundheitssystem bemerkbar machen.
Ob die HTA-Verordnung 2021/2282 für Entwickler von neuen Gesundheitstechnologien am Ende jedoch mehr oder weniger Aufwand bedeutet, wird wohl die Zeit zeigen.
Lassen Sie sich von unseren Expert:innen für klinische Bewertungen zeigen, wie Sie bestehende HTAs bei Ihrer klinischen Bewertung nutzen, damit Sie im Idealfall klinische Prüfungen vermeiden und auf diese Weise ihre Produkte deutlich schneller vermarkten können. Melden Sie sich gerne bei uns.


