
So stellen Sie sicher, dass Sie Ihre Inhouse-IVD (auch Laboratory Developed Tests, LDT genannt) auch unter der nun gültigen IVDR noch anbieten können. Sie haben drei Möglichkeiten, um Rechtsstreitigkeiten zu vermeiden.
Inhouse-IVD zählen zu den in-vitro-diagnostischen Tests. Gelten für diese Art von Produkten auch regulatorische Anforderungen wie die IVDR? Dieser Artikel verschafft nicht nur medizinischen Laboren einen Überblick.

1. Weshalb Sie Ihr Test-Angebot gegebenenfalls reduzieren müssen
Der EU-Gesetzgeber reguliert mit der EU-Verordnung 2017/746 über In-vitro-Diagnostika (IVDR) erstmals Inhouse-IVD. Damit betreffen die in der IVDR gestellten Anforderungen auch Labore, die eigenentwickelte Tests anbieten.
Die meisten dieser Forderungen sind zwar nicht neu, aber die zusätzlichen Anforderungen führen dazu, dass medizinische Labore bestimmte Inhouse-Tests nicht mehr anbieten dürfen. Dies kann nämlich der Fall sein, wenn ein Hersteller ein vergleichbares CE-IVD-Produkt im EU-Markt anbietet.
Was das für Sie bedeutet und wie Sie diese Problematik umgehen, erfahren Sie in diesem Artikel. Doch widmen wir uns zunächst den Grundlagen.
2. Inhouse-IVD: Was ist das überhaupt?
a) Begriffsdefinitionen
Die IVDR definiert weder Inhouse-IVD noch LDT unter den Begriffsbestimmungen in Artikel 2, nennt aber in Artikel 5 folgende Beschreibung:
Das Guidance Dokument MDCG 2023-1 der Medical Device Coordination Group benennt entsprechende Tests als „In-house device“:
Die US-amerikanische Zulassungsbehörde FDA definiert den Begriff „Laboratory Developed Tests“ wie folgt:
b) Abgrenzung zu Medizinprodukten aus Eigenherstellung
Inhouse-IVD, also In-vitro-Diagnostika aus Eigenherstellung, sind wiederum von den Medizinprodukten aus Eigenherstellung zu unterscheiden. Bei einem Medizinprodukt aus Eigenherstellung würde man von einer Eigenherstellung gemäß Artikel 5 (5) MDR sprechen.
c) Wo Inhouse-IVD verwendet werden
Medizinische Labore untersuchen Proben, die vom menschlichen Körper stammen: Gewebe und Körperflüssigkeiten wie Blut, Urin, Liquor usw. Um aus diesen Proben Diagnosen abzuleiten bzw. für die Diagnose relevante Informationen zu gewinnen, nutzen Labore
- kommerzielle In-vitro-Diagnostika (IVD) oder
- nicht CE-markierte, modifizierte oder eigenentwickelte Tests, die Inhouse-IVD.
Wo CE-IVD-Produkte verwendet werden, können unter bestimmten Umständen auch Inhouse-IVD eingesetzt werden. Die IVDR beschränkt den Einsatz von Inhouse-IVD jedoch auf sogenannte Gesundheitseinrichtungen und definiert diese wie folgt:
Medizinische Labore versorgen Patienten bzw. ihre behandelnden Ärzte mit medizinischen Informationen für die Diagnose. Darum verstehen wir entsprechend der Definition medizinische Labore als Gesundheitseinrichtungen.
Jede IVD-Untersuchung in einem medizinischen Labor beginnt damit, dass der Patientin oder dem Patienten eine Probe entnommen wird. Sie endet im Idealfall mit einem Ergebnis, das für die weitere Behandlung nützlich ist. Abb. 2 stellt die üblichen Schritte einer IVD-Untersuchung von der Probennahme bis zum Ergebnis dar. Hinter all diesen Schritten können sich neben CE-IVD-Produkten auch Inhouse-IVD verbergen.

d) Kriterien, die einen Test zum Inhouse-IVD machen
Ein Inhouse-IVD ist somit auch meist kein eigenentwickeltes Gerät. Es ist vielmehr ein Verfahren, das eigenentwickelte Produkte in Teilschritten der Untersuchung verwendet. Für Patienten und behandelnde Ärzte ist es in der Regel nicht ersichtlich, ob die Untersuchung mit einem Inhouse-IVD oder einem CE-IVD-Produkt durchgeführt wird (siehe Abb. 3).
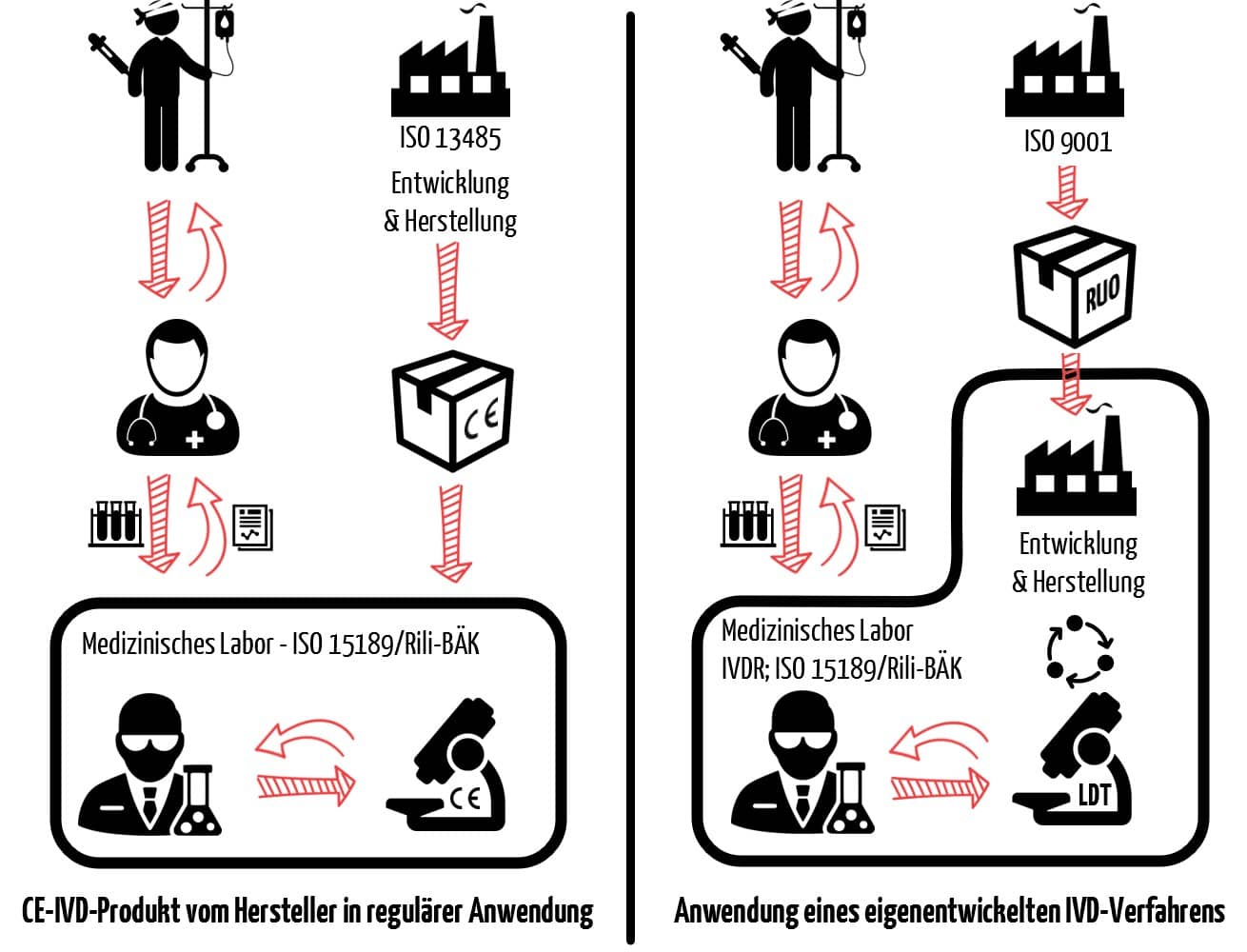
Ein medizinisches Labor wird zum Hersteller eines Inhouse-IVD, wenn es
- eigenentwickelte Produkte oder Verfahren verwendet,
- Nicht-IVD-Produkte im diagnostischen Verfahren verwendet (z. B. for research use only, RUO),
- Produkte oder Verfahren kombiniert, die nicht für die Kombination bestimmt sind, oder
- ein CE-IVD-Produkt außerhalb der Zweckbestimmung einsetzt, z. B. wenn Sie
- das Produkt zweckentfremden (off-label use) oder
- das Produkt abändern (hierzu zählt ggf. auch schon, wenn Sie von der Gebrauchsanweisung herkömmlicher IVD abweichen).
Wenn ein oder mehrere dieser Kriterien auf Ihre Tests zutreffen, gelten die im folgenden Kapitel gelisteten gesetzlichen Anforderungen in vollem Umfang.
3. Gesetzliche Anforderungen
a) Bisherige Anforderungen an Inhouse-IVD in Deutschland
Die Inbetriebnahme von In-vitro-Diagnostika aus Eigenherstellung war gemäß Medizinproduktegesetz (MPG) § 12 nur gestattet, wenn die grundlegenden Anforderungen der EU-Richtlinie 98/79/EG über In-vitro-Diagnostika (IVDD) erfüllt sind. Das bedeutet: Wenn Sie Ihr Inhouse-IVD bereits vor dem 26. Mai 2022 angeboten haben, müssen Sie für diesen den Anhang I der IVDD erfüllt haben.
Medizinische Labore müssen zudem die Richtlinie der Bundesärztekammer zur Qualitätssicherung laboratoriumsmedizinischer Untersuchungen, kurz Rili-BÄK, einhalten. Dies ist in der Medizinprodukte-Betreiberverordnung (MPBetreibV) festgelegt. Dies hat sich mit Gültigkeit der IVDR auch nicht geändert.
b) Anforderungen der IVDR an Inhouse-IVD
Die IVDR möchte „hausinterne“ Produkte weiterhin ohne Beteiligung einer Benannten Stelle und ohne eine CE-Kennzeichnung erlauben (Erwägungsgründe 28 und 29 im Vorwort der IVDR). Die IVDR formuliert in Artikel 5 (5) die Voraussetzungen, die für Tests zu erfüllen sind, die ausschließlich hausintern hergestellt und verwendet werden (dies schließt auch die remote-Anwendung von Inhouse-IVD Software ein; siehe MDCG 2023-1 3.2.2). Diese Voraussetzungen umfassen u.a.:
- Für alle Inhouse-IVD ist die Konformität mit den grundlegenden Sicherheits- und Leistungsanforderungen gemäß Anhang I der IVDR nachzuweisen.
- Die Herstellung und die Verwendung der Produkte erfolgen im Rahmen geeigneter Qualitätsmanagementsysteme.
- Weder die Rili-BÄK noch die ISO 15189 formulieren Anforderungen an die Entwicklung und die Herstellung von in-vitro diagnostischen Produkten. Um dieser Forderung nachzukommen, können Labore zum Beispiel die Anforderungen aus Kapitel 7 der ISO 13485 anwenden.
- Die Gesundheitseinrichtung liefert in ihrer Dokumentation eine Begründung dafür, dass die spezifischen Erfordernisse der Patientenzielgruppe nicht bzw. auf dem angezeigten Leistungsniveau nicht durch ein gleichartiges auf dem Markt befindliches Produkt befriedigt werden können.
- Dies ist sicherlich eine entscheidende neue Anforderung, weil damit viele Inhouse-IVD, für die es ein äquivalentes CE-IVD-Produkt am Markt gibt, keine Daseinsberechtigung mehr haben.
Weitere Anforderungen aus Artikel 5 (5), wie
- eine öffentlich zugänglichen Erklärung,
- die Überwachung der Produkte,
- das Verbot der Abgabe und
- das Verbot der Produktion im industriellen Maßstab
wurden bereits durch das MPG und den zugehörigen Verordnungen verlangt.
Sollten Sie feststellen, dass Sie eine der Anforderungen noch nicht erfüllt haben oder sollten Sie Fragen haben, kontaktieren Sie uns. Das Johner Institut unterstützt medizinische Labore bei der Umsetzung aller Anforderungen der IVDR.
c) Anforderungen der FDA an Laboratory Developed Tests
An dieser Stelle lohnt sich ein Blick über den großen Teich. Seit spätestens 2010 macht sich die FDA Gedanken zur Regulierung von LDT.
Die FDA sieht es demnach als problematisch an, dass ein Test, der von einem IVD-Hersteller entwickelt wurde, anders reguliert wird als ein identischer Test, den ein Labor entwickelt hat. Damit würde der risikobasierte Ansatz ausgehebelt. Die Überwachung solle abhängig vom Risiko erfolgen und nicht abhängig davon, wer den Test entwickelt.
Am 29. April 2024 veröffentlichte die FDA schließlich eine final rule, um die Sicherheit von LDTs zu gewährleisten. Sie ist sich der Risiken durch falsche oder ungeeignete Behandlung von Patienten bewusst, die durch nicht ausreichend kontrollierte LDTs entstehen können. Die Regel stellt klar, dass auch LDTs als IVD-Produkte unter den FD&C Act fallen. Die neuen Regeln werden über einen Zeitraum von vier Jahren schrittweise gültig und die bisherige zurückhaltende Überwachung beenden.
Ausnahmen
Einige LDTs sind von der Überwachung durch die FDA auszunehmen. Dazu zählen zum Beispiel
- Sehr simple LDTs ohne Automatisierung („1976 type LDTs“),
- HLA tests for transplantation,
- Forensic tests,
- DoD and VHA LDTs
Seltene Krankheiten werden nicht per se ausgenommen. Die FDA behält sich aber einen Ermessensspielraum und ggf. weitere Anpassungen ihrer Regeln vor.
d) Zusammenfassung der gesetzlichen Anforderungen
Nahezu alle Anforderungen der IVDR existierten bereits so oder in ähnlicher Form durch das MPG und die zugehörigen Verordnungen.
Der Anhang I der IVDR ist im Vergleich zum Anhang I der IVDD zwar länger geworden, aber die zentralen Elemente sind weiterhin
- das Risikomanagement,
- ein sicheres und leistungsstarkes Produktdesign,
- die Gebrauchstauglichkeit,
- die Leistungsbewertung und
- die Software Lebenszyklus-Prozesse.
Eine wesentliche neue Anforderung ist die IT-Security.
Ausnahmen von der Überwachung für bestimmte Inhouse-IVD (z. B. Inhouse-IVD zur Diagnose seltener Krankheiten), wie die FDA sie praktiziert, sind für den Europäischen Markt derzeit nicht bekannt.
Dr. Grömminger erläutert im Gespräch mit Professor Johner, was die Labore und Hersteller machen sollten und welche Auswirkungen die Verordnung auf die Geschäftsmodelle hat.
Diese und weitere Podcast-Episoden finden Sie auch hier.

4. Übergangsfristen
Im Januar 2022 wurden neue Übergangsfristen in die IVDR aufgenommen, die auch die Anforderungen aus Artikel 5(5) adressieren und somit die Inhouse-IVD betreffen. Im Juni 2024 wurde die Übergangsfrist für Artikel 5(5) d) nochmals angepasst.
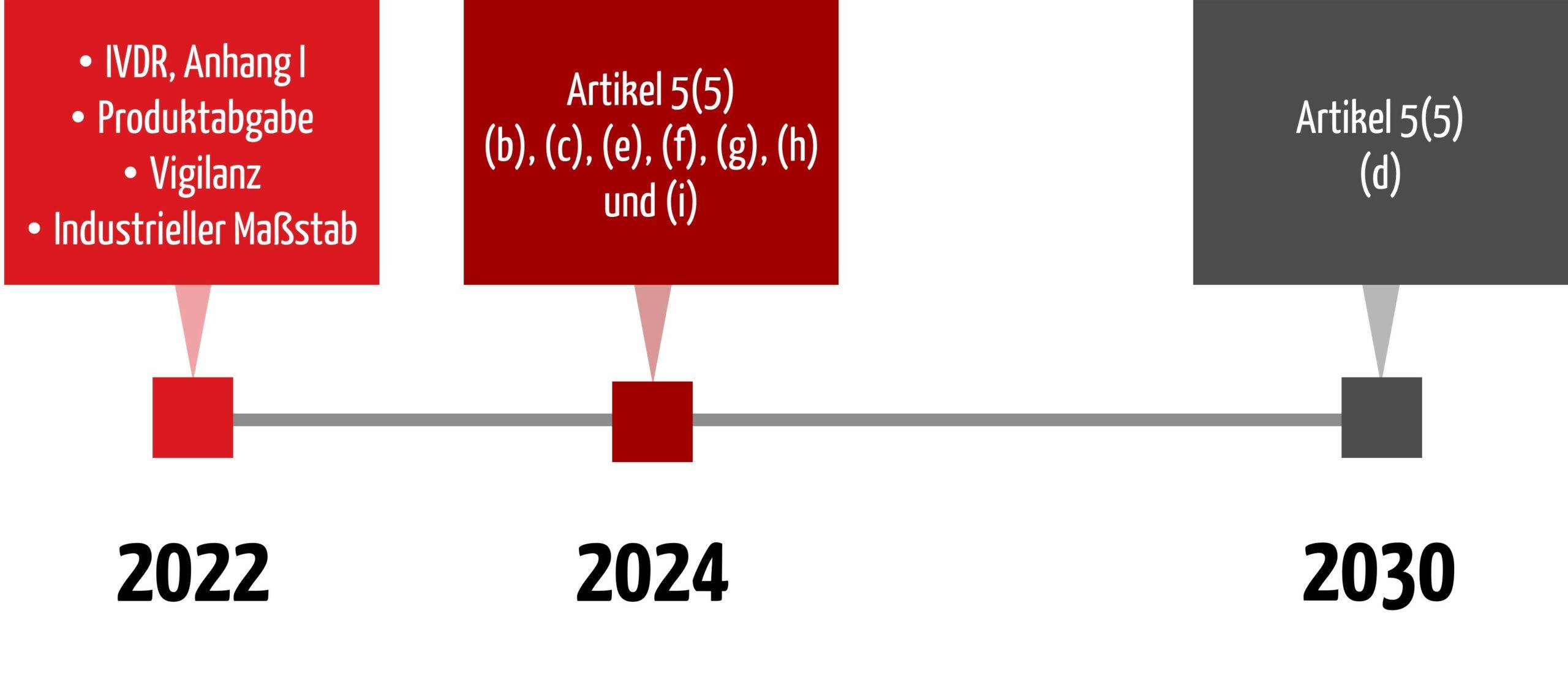
Wichtig
Die Anforderung nach Konformität mit Anhang I (Artikel 5 (5) Satz 1) besteht unter anderem seit dem 26. Mai 2022 und wurde nicht verschoben.
5. Überwachung durch Behörden
In Deutschland sind die jeweiligen Landesbehörden für die Überwachung der Labore auf Einhaltung der IVDR zuständig. Sie prüfen zudem, ob Labore die MPBetreibV einhalten, und damit auch die Einhaltung der Rili-BÄK. Im Falle einer Akkreditierung Ihres Labors gemäß ISO 15189 ist die DAkkS für die Überwachung der Umsetzung der Norm zuständig.
Die Rechte der Mitgliedsstaaten bei der Überwachung von Gesundheitseinrichtungen sind in IVDR Artikel 5 (5) Unterabsatz 2 explizit erwähnt.
MDCG 2023-1 nennt in Kapitel 3.7 Informationen, die von den Überwachungsbehörden eingesehen werden können.
6. Fazit
a) Die IVDR stellt konkrete Forderungen an Labore
Obwohl bereits das MPG die Erfüllung der grundlegenden Anforderungen des Anhangs I der IVDD forderte, erreichte man keine flächendeckende Einhaltung dieser Vorgaben.
Die IVDR nimmt Labore nun europaweit einheitlich in die Pflicht.
- Die eigenen Inhouse-IVD müssen Labore (weitgehend) konform zu den Anforderungen der IVDR entwickeln, herstellen und überwachen.
- Sie sollen bevorzugt kommerziell verfügbare IVD nutzen und können eigene Inhouse-IVD nur dann anbieten, wenn äquivalente Produkte auf dem Markt nicht die erforderliche Leistung bieten.
Die bisherige Praxis war auch schwer nachvollziehbar: Weshalb sollten die regulatorischen Anforderungen an einen Labortest davon abhängen, ob er durch ein Labor oder einen IVD-Hersteller entwickelt wurde?
Viele medizinische Labore sind sich der neuen Anforderungen nicht vollumfänglich bewusst. Es wird höchste Zeit, sich eingehend damit zu beschäftigen. Ein härterer Wettbewerb zwischen den Laboren sowie Rechtsstreitigkeiten mit IVD-Herstellern sind zu erwarten.
b) Ihre nächsten Schritte als medizinisches Labor
- Priorisieren Sie die Erfüllung der Anforderungen aus Anhang I und setzen diese JETZT um, sofern noch nicht geschehen.
- Erweitern Sie Ihr Qualitätsmanagementsystem um die geforderten Punkte und erzeugen Sie die daraus abgeleitete Dokumentation bis 2024.
- Erfüllen Sie die zusätzlichen Anforderungen des Artikel 5 (5) der IVDR.
- Prüfen Sie den Markt und suchen Sie nach Produkten, deren Leistungsversprechen für dieselbe Patientenzielgruppe ihrem Inhouse-IVD entsprechen.
Es gibt kein solches Produkt am Markt? Glückwunsch, dann dürfen Sie Ihr Inhouse-IVD über 2028 hinaus anbieten!
Gibt es jedoch ein äquivalentes Produkt auf dem gleichen Leistungsniveau im Markt, sollten Sie sich eine Strategie überlegen. Das Johner Institut unterstützt Sie dabei gerne.
Treffen Sie eine Entscheidung:
- Test nicht mehr anbieten
- Zugelassenen CE-IVD-Test kaufen und verwenden
- Eigenen Test als CE-IVD in Verkehr bringen
Bei der Entscheidung „Test als CE-IVD in Verkehr bringen“:
- Implementieren Sie ein QMS nach ISO 13485.
- Suchen Sie sich eine Benannte Stelle.
- Erfüllen Sie die weiteren Anforderungen der IVDR (jeweils abhängig von der Art und der Risikoklasse des Produkts), erstellen Sie die produktspezifische technische Dokumentation und erklären Sie die Konformität.
Über das Starter-Kit des Johner Instituts erhalten Sie Ihre kostenlose Version der Checkliste für den Anhang I der IVDR.
Das IVD-Team unterstützt Sie gerne bei der Einhaltung der IVDR-Vorgaben für Inhouse-IVD.
Im Seminar IVDR für medizinische Labore gibt Ihnen unser Laborexperte Ulrich Hafen einen umfangreichen Überblick über die Anforderungen der IVDR an die Verwendung von Inhouse-IVD. Sie lernen, wie Sie Ihre bestehenden Inhouse-Tests konform betreiben und sind nach Abschluss des Seminars in der Lage, neue Inhouse-IVD anhand eines erarbeiteten Fahrplans effizient IVDR-konform zu dokumentieren.
Wenn Sie bereits wissen, dass Sie künftig zum CE-IVD-Hersteller und Inverkehrbringer werden wollen, empfehlen wir Ihnen das Seminar Technische Dokumentation nach IVDR.
Änderungshistorie
- 2024-06-04:
- Überarbeitung von Kapitel 3 c) zu den neuen Regeln der FDA an LDTs.
- Anpassung von Kapitel 4 an neue Übergangsfristen.
- 2023-01-12:
- Anpassung der Definition „In-house device“ entsprechend der neuen MDCG 2023-1.
- Verweis auf MDCG 2023-1 und leichte Anpassungen in Abschnitt 3. b)
- Verweis auf MDCG 2023-1 in Abschnitt 5.
- 2022-12-09: Hinweise zur Überarbeitung von ISO 15189 ergänzt
- 2022-06-08:
- Umstellungen aufgrund der gültig gewordenen IVDR
- „LDT“ durch „In-house IVD“ ersetzt, dem offiziellen Wording der EU
- Anpassungen aufgrund der Übergangsfristen
- Entfernung des Abschnitts 3 e) „Auswirkungen bei anhaltender Corona-Pandemie“
- 2021-04-22: Abschnitt 3. e) zu den Auswirkungen bei anhaltender Corona-Pandemie eingefügt. Redaktionelle Änderungen.
- 2021-12-01: Korrekturen, Anpassungen von Verlinkungen und redaktionelle Änderungen im gesamten Artikel. Anpassung der Abschnitte
- 2. a): Überarbeitung der Definitionen
- 3. b): Umstrukturiert
- 4.: Neuen Abschnitt zu Übergangsfristen eingefügt
- 6. b): Anpassung an eventuelle neue Übergangsfristen



Hallo Herr Johner,
eine sehr nützliche Zusammenfassung, die ich gerne verlinken werde. Werden Sie den Artikel auch ins Englische übersetzen? Das wäre super!
Viele Grüße
Ben Liesfeld
Sehr geehrter Herr Liesfeld,
danke für das positive Feedback, über das ich mich freue!
Der Artikel ist bereits übersetzt. Sie finden ihn auf unserer Englischen Seite:
https://johner-institute.com/articles/regulatory-affairs/and-more/laboratory-developed-tests/
Ich ergänze auf der deutschen Seite den Link. Danke für die Anregung!
Viele Grüße, Christian Johner
Ein toller überblick, wenn auch mit keinem schönen Inhalt aber so weiß man gut was auf uns zukommt. Vielen Dank, hatte lange nach so einem Artikel gesucht.
Sehr geehrter Herr Hafen!
Ist eine Kombination von zwei CE-markierten IVDs (z.B. ein ELISA Automat und ein ELISA Kit), wenn die Kombination nicht spezifisch von einem Hersteller validiert wurde, auch ein LTD?
Wenn ja, wer ist verantwortlich für die Validierung?
Der Hersteller des ELISA Automats müsste ja eine Einschränkung in der Gebrauchsanweisung angeben, nachdem der Automat eindeutig zur Verwendung in Kombination mit anderen Produkten Ausrüstungen bestimmt ist?
Oder ist es der Anwender, der die beiden Produkte kombiniert? Braucht es dazu die ganze Palette an Anforderungen oder reicht in dem Fall eine „kleine“ Validierung der Kombination?
Ich danke Ihnen sehr für Ihre Hilfe!
Sehr geehrte Frau Mitterhuber,
das kommt ganz auf die Zweckbestimmungen von Gerät und Kit an. Wenn Das ELISA-Kit in seiner Gebrauchsanweisung z. B. keinen definierten Reader vorschreibt, sondern technische Anforderungen an einen Reader stellt und der CE-IVD Reader die Anforderungen erfüllt und wiederum nicht auf bestimmte Kits in seiner Gebrauchsanweisung beschränkt ist, wäre eine Verwendung möglich, ohne eine Eigenherstellung vorgenommen zu haben.
Ist dies nicht der Fall, stellen Sie bei Kombination ein LDT her und sind selbst verantwortlich für die Einhaltung der in IVDR Art. 5 (5) gestellten Anforderungen zu denen die Leistungsbewertung (Validierung) gehört sowie auch Anhang I wobei hier Abschnitt 13.1 hervorzuheben ist.
Bei der Leistungsbewertung können Sie natürlich auf bereits durch den Inverkehrbringer generierte Leistungsdaten, die nicht durch Ihre Anwendung beeinflusst werden, verweisen. Es sind die kritischen Einflussgrößen zu bestimmen und diese zu validieren.
Beste Grüße,
Ulrich Hafen
Sehr geehrter Herr Hafen,
vielen Dank für Ihre fundierte Antwort!
Wie verhält es sich mit einer Kombination aus RUO-Gerät und CE-markiertem Test, wenn die Kombination vom Hersteller validiert und in der Gebrauchsanweisung offiziell freigegeben wird?
Handelt es sich um ein LDT mit allen Anforderungen der IVDR?
Vielen Dank und beste Grüße!
Liebe Frau Mitterhuber,
solch eine Kombination kann nicht von einem Inverkehrbringer eines CE-IVD als für die Diagnostik validiert und „freigegeben“ existieren, da ein RUO produkt, wie der Name „Research Use Only“ schon sagt, nur für die Forschung und nicht für Diagnostik bestimmt ist. Inverkehrbringer von CE-IVD müssen jedoch in der Gebrauchsanweisung die spezifikationen der Produkte des allgemeinen Laborbedarfs angeben, welcher benötigt wird, um das Produkt sicher anzuwenden. Wenn Ihr allgemeiner Laborbedarf den Spezifikationen des Inverkehrbringers entspricht und die Anforderungen an Wartung und Pflege des allgemeinen Bedarfs eingehalten werden, können sie die entsprechenden Produkte des allgemeinen Bedarfs nutzen, ohne dass ein Inhouse-IVD hergestellt wurde.
Hallo Herr Hafen,
tolle Übersicht, vielen Dank! Heißt das somit, dass man in der EU kein Produkt unter der IVDR als LDT verkaufen kann, wenn es ein äquivalentes Produkt gibt, aber in den USA nicht eine solche Regel gibt? Wenn es in USA bereits das Produkt als LDT gibt, bei dem sich der Hersteller nun doch entscheidet dies auf offiziellem Weg bei der FDA genehmigen zu lassen, ändert sich etwas für die LDT oder dürfen parallel FDA genehmigter Test und LDT auf dem Markt existieren?
Das wäre somit der Unterschied zw. EU und USA, wenn das richtig ist.
Vielen Dank,
Stefanie
Liebe Stefanie,
zuerst möchte ich Sie um Entschuldigung bitten, da Sie auf Grund meines Urlaubs so lange auf eine Antwort warten mussten.
Die Anforderung auf die Sie anspielen ist IVDR Artikel 5(5) d). Diese Anforderung besagt, dass LDT nicht mehr angeboten werden dürfen, sofern es ein äquivalentes Produkt auf dem gleichen Leistungsniveau gibt. Das bezieht sich ausschließlich auf in der EU in Verkehr gebrachte Produkte. Demnach ist es möglich in der EU einen LDT auch noch nach 2028 zu betreiben, sofern es kein äquivalentes CE-markiertes Produkt gibt, unabhängig davon, ob in den USA ein äquivalentes Produkt zugelassen wurde.
Ich hoffe, ich konnte Ihre Frage zu Ihrer Zufriedenheit beantworten. Falls nicht, nutzen Sie gerne auch unser kostenloses MicroConsulting. Ich freue mich auch Sie als Teilnehmerin meines Seminars begrüßen zu dürfen in dem wir solchen Fragestellungen zu Grunde gehen.
Liebe Grüße,
Ulrich Hafen
Guten Tag Herr Hafen,
ich weiss nicht, ob sie der richtige Adressat für meine Frage sind, aber ich versuche es trotzdem einmal. Ist Ihnen eine rechtlich verbindliche Quelle bekannt, die klar aussagt, dass im medizinischen Labor ausschließlich Produkte (Medizinprodukte/In-vitro-Diagnostik-Produkte)mit einem CE-Kennzeichen verwendet werden dürfen. Ich meine hiermit auch Produkte wie bspw. Probenbehältnisse für Proben aus dem menschlichen Körper, Blutentnahmeröhren und all der Kram, der üblicherweise unter Praxisbedarf geführt wird. Das gilt natürlich gleichermassen für die großen Laborautomaten oder Untersuchungsgeräte.
Bis jetzt habe ich dazu nur im Sozialgesetzbuch eine nicht ganz eindeutige Quelle gefunden, die sich aber auf die Erstattung von Leistungen durch die Krankenversicherungen bezieht. aber gilt das auch für Praxsbedarf? Ich freue mich über eine Rückmeldung.
Sehr geehrter Herr Lichtenthal,
Sie sind mit Ihrer Frage goldrichtig bei uns/mir.
Eine rechtlich verbindliche Quelle, die die Aussage trifft, dass in einem diagnostischen Untersuchungsverfahren ausschließlich CE-gekennzeichnete Produkte zum Einsatz kommen müssen, gibt es NICHT.
Und auch aus gutem Grund nicht. Würde es eine solche Vorgabe geben, hätten wir in der EU keine labormedizinische Versorgung mehr, da wir es uns nicht mehr leisten könnten.
Wie in meinem Artikel beschrieben (Kapitel 3. b)), erlaubt die IVDR in Artikel 5 (5) den Betrieb von In-house Produkten, die kein CE-Zeichen tragen. Darüber hinaus ist es weiterhin gestattet allgemeinen Laborbedarf im Rahmen der Anwendung von CE-IVD zu nutzen, sofern die Produkte des allgemeinen Bedarfs die Anforderungen des CE-IVDs erfüllen. Die Anforderung an solche Produkte des allgemeinen Bedarfs müssen in der Gebrauchsanweisung des CE-IVD dokumentiert sein (IVDR Anhang I 20.4.1. i) und j)) und natürlich vom Labor erfüllt werden.
Da Sie Probenbehältnisse explizit ansprechen – Vorsicht: Probenbehältnisse, also auch Blutentnahmeröhrchen, sind IVD (siehe IVDR Artikel 2 2.) und werden nach Anhang VIII der Klasse A zugeordnet. Wird also für den Transport von Blut für eine nachfolgende diagnostische Untersuchung ein Röhrchen ohne CE-Zeichen genutzt, wäre das nicht konform, es sei denn das Röhrchen des allgemeinen Bedarfs ist als Teil eines In-house IVD leistungsbewertet.
Freundliche Grüße,
Ulrich Hafen
Guten Morgen Herr Hafen,
vielen Dank für ihre Antwort. Dann ist die CE-Kennzeichnung also verpflichtend für alle Probenbehältnisse (IVD Klasse A) zum Transport und zur Aufbewahrung von Untersuchungsgut aus dem menschlichen Körper für diagnostische Untersuchungen, was auch Urinbecher, Biopsie-Einbettkassetten etc. umfassen würde?
Lieber Herr Lichtenthal,
wenn das entsprechende Behältnis die Definition von „Probenbehältnis“ in IVDR Artikel 2 3. erfüllt, also eine entsprechende Zweckbestimmung besitzt um „aus dem menschlichen Körper stammende Proben unmittelbar nach ihrer Entnahme aufzunehmen und im Hinblick auf eine In-vitro-Untersuchung aufzubewahren“, ist das Probenbehältnis ein IVD und benötigt eine CE-Kennzeichnung.
Ob das auf Biopsie-Einbettkassetten zutrifft, hängt von deren Zweckbestimmung ab. Werden diese genutzt zur unmittelbaren Aufnahme der Probe nach der Entnahme, oder wird dafür doch ein anderes Probenbehältnis genutzt und die Überführung der Probe in die Biopsie-Einbettkassette findet erst nach Eintreffen der Probe im Labor statt.
Sollten Sie weiteren und intensiveren Bedarf an Austausch haben, freue ich mich, wenn Sie sich über unser Kontaktformular an uns wenden und wir Sie in einem gemeinsamen Projekt unterstützen dürfen.
Herzliche Grüße,
Ulrich Hafen
Sehr geehrter Herr Hafen
Laut IVDD waren sogenannte Middleware – oder Expertensystemen von der Diagnostika Direktive ausgeschlossen. D.h. Softwarelösungen, die nur Labor-Ergebnisse und Patientendaten miteinander vergleicht und daraus sogenannte Therapieempfehlungen erstellen, unterlagen nicht der IVDD.
Hat sich das durch die IVD-R geändert?
Sehr geehrter Herr Vitzthum,
ich danke Ihnen sehr für Ihre spannende Frage.
Generell war Software, zumindest namentlich, nicht Teil der IVD Definition in der IVDD. Gleichwohl gab es natürlich CE-IVD Software unter der Richtlinie. Aber das nur nebenbei.
In der IVDR ist nun der Begriff „Software“ auch in der Definition eines IVD zu finden. Somit ist jegliche Software, die der Definition eines IVD in Artikel 2.2. der IVDR entspricht, auch ein IVD. Das betrifft natürlich auch (eigenentwickelte) In-house Software. Ob dies zutrifft ist abhängig von der Zweckbestimmung der Software. Eine Sonderregelung für „Expertensysteme“, wie noch in MEDDEV 2.1/6, gibt es in der IVDR und in der MDCG 2019-11 nicht mehr.
Schreiben Sie also für Ihr Expertensystem eine ausführliche Zweckbestimmung und gleichen diese mit der Defnition eines IVDs ab. Nutzen Sie hierzu auch MDCG 2019-11. Gerade bei Therapieempfehlungen kann es gut sein, dass die Software ein IVD ist.
Ich empfehle Ihnen dringend auch einen Blick in folgende unserer Artikel:
https://www.johner-institut.de/blog/regulatory-affairs/ivd-software/ insbesondere 1. b)
https://www.johner-institut.de/blog/iec-62304-medizinische-software/decision-support-systeme-medizinprodukt/
Herzliche Grüße,
Ulrich Hafen
Sehr geehrter Herr Hafen,
auch wenn Ihr Artikel jetzt schon älter ist, verliert er nicht an Aktualität, vielen Dank für die sehr klare Darstellung.
Mir ist nun untergekommen, dass überwachende Behörden (Einhaltung der RiliBäk) fordern, dass zur Erfüllung der Anforderungen für die Validierung von LDTs eine vorhandenenr Akkreditierung nach DIN EN ISO 15189 Voraussetzung ist. Wenn ich Sie richtig verstehe, muss „nur“ ein entsprechendes QMS, das die Einhaltung der entsprechenden IVD-R Anforderungen sicherstellt, vorhanden sein. Eine Akkreditierung für das Angebot von diagnostischen LDTs ist aber nicht zwingend notwendig.
Ich freue mich über eine Klarstellung.
Vielen Dank!
Liebe Frau Hock,
ich danke Ihnen sehr für Ihr Lob und versichere Ihnen, dass wir unsere Artikel auch fortlaufend aktuell halten, sofern Anpassungsbedarf identifiziert wird.
Zu Ihrer Frage: Sie haben absolut recht. Eine Akkreditierung nach ISO 15189 ist zur konformen Entwicklung und Betrieb von Inouse IVD NICHT erforderlich. Die IVDR schreibt in Artikel 5(5)c) „das Labor der Gesundheitseinrichtung entspricht der Norm EN ISO 15189 oder gegebenenfalls nationalen Vorschriften“. Es „entspricht“ der 15189, nicht „es muss akkreditiert sein nach“. Dies wird auch bestätigt durch 1. MDCG 2023-1. Dort steht: „Compliance with the standard EN ISO 15189 may be understood as accreditation to the standard or other means of compliance.“ 2. gibt es in Deutschland keine Zwangsakkreditierung. 3. gibt es keine nationale Anforderung nach einem ISO 15189 System, aber dafür nach Einhaltung der Rili-BÄK Teil A in der MPbetreibV.
Einrichtungen mit Inhouse IVD benötigen das sog. erweiterte Managementsystem, bestehend aus Rili-Bäk/ISO 15189 (beide sind relativ ähnlich) zuzüglich erweiterungen zur Entwicklung, wie in ISO ISO 13485, Kapitel 7 zu finden.
Die Voraussetzung einer Akkreditierung nach ISO 15189 durch eine Landesbehörde ist nicht gerechtfertigt.
Lieber Ulrich, weißt Du ob eine Registrierung für In-House IVDs beim BfArM oder auf EU-Ebene geplant ist? Oder gilt die Registrierungspflicht ausschließlich für Medizinprodukte und IVDs, die In Verkehr gebracht werden?
Liebe Grüße, Sebastian
Hi Sebastian, schön von Dir zu lesen!
bisher ist uns keinerlei Registrierungspflicht für Inhouse-IVD, weder auf EU-Ebene, noch auf nationaler deutscher Ebene bekannt (für die Schweiz ist solch eine Registrierungspflicht in Art. 10 IvDV festgeschrieben).
Im gegenteil, eine kürzliche Korrespondenz von uns mit dem BfArM bezüglich Anzeigepflicht von Leistungsstudien von Inhouse-IVD ergab überraschenderweise, dass diese auch nicht beim BfArM anzuzeigen sind.
Herzliche Grüße,
Ulrich