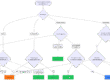
PDMS steht für Patientendatenmanagementsystem. Diese klinischen Informationssysteme finden sich typischerweise in Krankenhäusern, v. a. in den Abteilungen, die Patienten intensivmedizinisch behandeln.
Durch die Förderungen des Krankenhaus-Zukunftsgesetzes (KHZG) erleben die PMDS einen neuen Boom.
Dieser Artikel
- verschafft einen Überblick über PDMS,
- nimmt eine regulatorische Einordnung vor (z. B. Qualifizierung als Medizinprodukt),
- und gibt Hilfestellungen für das Risikomanagement.

1. PMDS: Funktionalitäten und Anforderungen
Patientendatenmanagementsysteme (PDMS) müssen viele Anforderungen erfüllen und Funktionen anbieten:
- Datenerfassung und -integration
- Patientenüberwachung
- Klinische Entscheidungshilfe
- Berichterstattung und Analyse
Zum einen verfügen PDMS über Funktionalitäten eines „normalen“ klinischen Arbeitsplatzes, wie die Verwaltung von Patientenstammdaten, Anamnesen, Diagnosen, Befunden (z. B. Labor), Medikation und anderen Therapien.
Zum anderen zeichnen sie sich durch eine enge Verzahnung mit der Medizintechnik aus: Sie zeichnen Daten von Patientenmonitoren auf (z. B. Blutdruck, Puls, Sauerstoffsättigung) und auch von Beatmungsgeräten.
1.1 Datenerfassung und -integration
Ein PDMS muss in der Lage sein, Patientendaten aus verschiedenen Quellen zu sammeln und zu integrieren, z. B. von Patientenmonitoren, Beatmungsgeräten, aus elektronischen Gesundheitsakten (EHR), Laborinformationssystemen, Radiologie-Informationssystemen (RIS) und anderen klinischen Systemen.
Zu diesen Daten zählen oft demografische Daten des Patienten, die Krankengeschichte, Testergebnisse, bildgebende Untersuchungen und Informationen zur Medikation sowie „Gerätedaten“ von z. B. Beatmungsgeräten.
1.2 Patientenüberwachung
Ein PDMS sollte eine Patientenüberwachung in Echtzeit anbieten, um Änderungen im Zustand des Patienten zu erkennen und die Pflegekräfte zu alarmieren. Dazu gehören die Überwachung der Vitalparameter, der Medikation, der Beatmung und anderer für den Gesundheitszustand des Patienten spezifischer Parameter.
1.3 Klinische Entscheidungshilfe
Die Anwender erwarten von einem PDMS, dass es bei klinischen Entscheidungen unterstützt. Das sind
- Entscheidungen über die Art der Patientenversorgung,
- Warnung vor Arzneimittelinteraktionen,
- Überwachung des Flüssigkeitshaushalts,
- Hilfe bei der Dosierungsberechnung,
- Diagnoseempfehlungen und
- die Priorisierung von Patienten.
1.4 Berichterstattung und Analyse
Bei PDMS ist eine fortschrittliche Berichterstattungs- und Analysefunktionen üblich. Diese dient den Krankenhäusern bei der
- Abrechnung (insbesondere Art und Dauer der Beatmung wirken sich stark auf die Vergütung aus),
- Bewertung der Patientenergebnisse,
- Verfolgung von Leistungskennzahlen und
- Ermittlung verbesserungswürdiger Bereiche.
Viele PDMS helfen, Berichte zu erzeugen über
- die demografischen Daten der Patienten,
- klinische Ergebnisse und Probleme sowie
- die finanzielle Leistung, einschließlich Möglichkeiten zur Verbesserung der Abrechnung.
2. Regulatorische Einordnung
2.1 Qualifizierung von PDMS als Medizinprodukte
2.1.1 PDMS als Dokumentationssystem
Klinische Informationssysteme, die ausschließlich der Dokumentation dienen (wie das viele Hersteller von ihren Krankenhaus-Informationssystemen behaupten), fallen nicht unter die Definition „Medizinprodukt“.
Ein System wird erst dann zum Medizinprodukt, wenn es der Behandlung oder Diagnose von Krankheiten und Verletzungen oder der Überwachung physiologischer Parameter dient.
Obwohl viele PDMS die Überwachung physiologischer Parameter erlauben, argumentieren dennoch einige Hersteller, dass diese Aufzeichnungen nur der Dokumentation dienen.
2.1.2 PDMS als Medizinprodukt
Wenn die Zweckbestimmung allerdings Berechnungen beinhaltet, z. B. den Flüssigkeitshaushalt des Patienten oder die Wechselwirkung oder Kontraindikationen von Medikamenten betreffend, lässt sich nicht mehr bestreiten, dass das PDMS der Therapie dient. Auch Alarmfunktionen dürften diesem Zweck dienen.
Damit ist das PDMS ein Medizinprodukt (üblicherweise eines der Klasse IIa oder IIb) und muss entsprechend der gesetzlichen Vorgaben entwickelt und betrieben werden.
2.2 Regulatorische Anforderungen an PDMS in Europa
Daher zählen die meisten PDMS inzwischen als Medizinprodukt. Folglich müssen die Hersteller die allgemeinen Sicherheits- und Leistungsanforderungen (GSPR) erfüllen und nachweisen. Die EU-Medizinprodukteverordnung MDR legt GSPR in Anhang I fest, u. a.:
- Software-Lebenszyklusprozesse (IEC 62304)
- Risikomanagement (ISO 14971) (siehe unten)
- IT-Sicherheits-Lebenszyklusprozesse (IEC 81001-5-1)
- Usability Engineering (IEC 62366-1)
- QM-System (ISO 13485)
Beachten Sie die Seite zu den klinischen Informationssystemen. Sie enthält weiterführende Links, z. B. zur Qualifizierung und Klassifizierung von Standalone-Software, zur Parametrierung klinischer Informationssysteme sowie zum Betrieb und zur Eigenherstellung von Informationssystemen.
Dieser Fachartikel beschreibt die 7 Schritte, die notwendig sind, um Medizinprodukte wie ein PDMS zu „zertifizieren“ und bis zum CE-Zeichen zu gelangen. Der Prozess von der Entscheidung bis zur „Zertifizierung“ und Konformitätserklärung dauert typischerweise zwischen 9 und 24 Monaten.
Darüber hinaus unterliegen Betreiber eines PDMS den Datenschutzanforderungen (z. B. Anforderungen der DSGVO). Diese zu erfüllen gelingt nur, wenn die Hersteller von PDMS die technischen Voraussetzungen dafür schaffen, u. a. die Möglichkeit, gezielt Daten zu löschen.
2.3) Regulatorische Anforderungen an PDMS in den USA
Seit dem 21st Century Cures Act zählen viele Software-Anwendungen nicht mehr als Medizinprodukt.
Zur Abgrenzung legt die FDA ein Guidance-Dokument vor, das auf die Charakteristiken und Klassifizierungen von Datenerfassungssystemen im Krankenhaus eingeht. Dazu zählen Medical Device Data Systems, Medical Image Storage Devices und Medical Image Communications Devices.
Das Team der Regulary Experts des Johner Instituts hilft Herstellern von PDMS, eine regulatorische Strategie festzulegen. Dazu zählt u. a., die Märkte und deren Reihenfolge festzulegen und die Zweckbestimmung so zu bestimmen, dass das PDMS wie gewünscht qualifiziert wird.
Melden Sie sich gleich, um in den nächsten Tagen einen kostenlosen und unverbindlichen Termin zu vereinbaren. Dann würden wir gemeinsam die ersten Schritte Ihrer regulatorischen Strategie gehen und damit die Voraussetzung für eine schnelle Zulassung schaffen.
Die Regeln zur Qualifizierung von Medizinprodukten unterscheiden sich in Europa und den USA. Aber die Anforderungen der FDA an Medizinprodukte (wenn diese als solche qualifiziert sind), sind vergleichbar mit den Anforderungen der MDR. Daher empfiehlt es sich, die Zulassungsunterlagen für beide Märkte gemeinsam zu erstellen.
Allerdings sollten Hersteller von PDMS in den USA weitere regulatorische Anforderungen beachten:
- HIPAA
Der Health Insurance Portability and Accountability Act (HIPAA) legt nationale Standards für den Schutz der Privatsphäre und die Sicherheit von Gesundheitsdaten fest. PDMS müssen die HIPAA-Vorschriften einhalten, um die Sicherheit und Vertraulichkeit der Patientendaten zu gewährleisten. - CMS
Die Centers for Medicare and Medicaid Services (CMS) verlangen von den Krankenhäusern die Einführung von elektronischen Patientenakten mit bestimmten Standards, um in den Genuss von Bonuszahlungen zu kommen. PDMS müssen diese Standards erfüllen, um die Voraussetzungen für CMS-Anreize zu erfüllen.
3. Risikomanagement bei PDMS
Die MDR verlangt ein akzeptables Nutzen-Risiko-Verhältnis. Das bedeutet, dass Hersteller von PDMS deren Nutzen quantifizieren sollten, um diesen gegen die Risiken abwägen zu können.
3.1 Den Nutzen eines PDMS bestimmen
Beispiele für den Nutzen eines PDMS sind:
- Die Dokumentation mittels PDMS ist vollständiger und korrekter als die manuelle Dokumentation.
- Fehlberechnungen und Fehlmedikationen sind unwahrscheinlicher.
- Patienten in kritischem Zustand werden schneller und präziser identifiziert.
- Das medizinische Personal wird entlastet.
Hersteller müssen den Nutzen, den sie in ihrer Risikomanagementakte beschreiben, auch in der klinischen Bewertung nachweisen. Das ist nicht immer einfach. Daher ist die Wahl der „medical claims“ entscheidend.
Die klinischen Expertinnen und Experten des Johner Instituts helfen dabei, diese Nutzenversprechen quantitativ zu beschreiben und „auditsicher“ nachzuweisen.
3.2 Typische Fehlerquellen kennen
Damit der Nutzen tatsächlich überwiegt und die Risiken so gering wie möglich sind, sind Hersteller verpflichtet, typische Fehler zu vermeiden oder zumindest zu minimieren.
Beispiele für diese Fehler sind:
- Software-Bugs, z. B. bei der Implementierung von Algorithmen
- Interoperabilitätsprobleme: Daten werden etwa nicht, nicht korrekt (z. B. falsche Einheit) oder nicht in der spezifizierten Zeit zwischen dem PDMS und der Medizintechnik oder anderen Produkten und Systemen übertragen. Nicht nur das PDMS, sondern auch die anderen Systeme und Geräte kommen als Fehlerquellen in Betracht.
- Fehlerhafte Konfigurationen des PMDS (auch durch die Betreiber)
- Probleme bei der Gebrauchstauglichkeit: Beispielsweise nehmen die Anwender Alarme nicht wahr, verwechseln Patienten, geben falsche Daten ein oder missverstehen Informationen.
- Mangelnde IT-Sicherheit der Komponenten und des Entwurfs (“Security by Design”)
Nutzen Sie die Taxonomie der ISO 25010 als Checkliste, um die Software-Qualitätseigenschaften Ihres PDMS zu prüfen.
3.3 Gefährdungen, Risiken und Schäden ermitteln und vermeiden
Durch diese Fehler können sich Gefährdungen ergeben, beispielsweise:
- Patienten werden nicht behandelt, weil ein Nutzer verbotenerweise nicht auf den Primäralarm eines medizintechnischen Geräts geachtet, sondern sich auf Informationen im PDMS verlassen hat.
- Patienten werden fehlerhaft ernährt bzw. mit Flüssigkeit versorgt, weil z. B. die Flüssigkeitsbilanz falsch berechnet wurde.
- Patienten erhalten falsche Medikamente oder falsche Dosen, weil die medizinische Dokumentation fehlerhaft ist.
- Ein Cyber-Angriff führt zum Ausfall des PDMS. Die PDMS werden nicht mehr adäquat überwacht, weshalb notwendige medizinische Interventionen unterbleiben.
Die Expertenteams für Risikomanagement und IT-Security des Johner Instituts haben für zahllose klinische Informationssysteme die Risiken identifiziert (auch durch Pen-Tests) und geholfen, diese Risiken zu beherrschen und Zulassungsunterlagen zu erstellen. Mit dieser Unterstützung gelingt es Ihnen, Ihre sicheren Produkte schnell und plangemäß zuzulassen und in den Markt zu bringen.
Melden Sie sich und nutzen Sie diesen Erfahrungsschatz. So kommen Sie schneller und zu niedrigeren Kosten ans Ziel.
4. Fazit und Zusammenfassung
Patientendatenmanagementsysteme (PDMS) sind aus den Krankenhäusern, insbesondere aus den Intensivstationen, nicht mehr wegzudenken. Ohne diese Systeme würde es für das klinische Personal schwierig, die Datenflut, die insbesondere durch die Medizingeräte entsteht, zu überblicken und die Patienten adäquat zu überwachen und zu behandeln.
Weil PDMS fast immer der Überwachung und Behandlung von Krankheiten und Verletzungen dienen, zählen sie als Medizinprodukte. Daher müssen die Hersteller von PDMS die gesetzlichen Anforderungen an Medizinprodukte erfüllen und auch weitere regulatorische Anforderungen beachten.
Die regulatorischen Anforderungen und der Umgang damit unterscheiden sich bei PDMS nur wenig von anderer SaMD (Software as Medical Device).
Allerdings sind die Risiken und der Nutzen eines PDMS spezifisch für diese Produktklasse. Die Risken hängen zudem von der konkreten Installation und den verbundenen Medizingeräten und Software-Anwendungen ab.
Unterstützung für Hersteller von PDMS
Beratung bei der regulatorischen klinischen Strategie
Ziehen Sie bei der Entwicklung eines PDMS unsere Experts rechtzeitig für die regulatorische und klinische Strategie hinzu. So sind Sie sicher, dass Sie
- Ihr Produkt korrekt qualifizieren, klassifizieren und gesetzeskonform bewerben,
- die regulatorischen Anforderungen ermitteln und
- diese möglichst schnell und einfach erfüllen, um sowohl unnötige Kosten als auch Ärger mit Behörden und Benannten Stellen zu vermeiden.
Unterstützung bei der Überführung von „Bestandsprodukten“
Wenn Ihre Produkte schon lange im Markt sind, müssen Sie fortlaufend den Stand der Technik ermitteln und erfüllen. Gerade beim Übergang der Zertifikate von MDD nach MDR tun Hersteller gut daran, mithilfe eines neutralen Blicks von außen die Konformität der Systeme zu prüfen oder wiederherzustellen.
Bestandsprodukte leiden in vielen Fällen unter einem Mangel an ausreichender IT-Sicherheit. Hier helfen wir durch nachträgliche Aktivitäten wie Threat Modeling oder Penetration-Tests.
Hilfe bei der Risikomanagementakte und der Post-Market Surveillance
Das Risikomanagement ist die zentrale regulatorische Anforderung. Die Dokumentation durch die eigene Überwachung nach dem Inverkehrbringen auf Stand zu halten, fällt vielen Herstellern erfahrungsgemäß schwer. Wir unterstützen Sie mit Rat und Tat durch ein Review Ihrer Risikomanagement-Akten oder durch unsere Postmarket-Surveillance-Dienstleistungen.
Prüfung des QM-Systems und Auditvorbereitung
Ein internes Produkt- oder Systemaudit durch unsere erfahrenen Auditoren, die auch bei Benannten Stellen gearbeitet haben, hilft Ihnen, Lücken frühzeitig noch vor einer Begutachtung durch die Benannte Stelle zu erkennen und zu schließen.