
Die EU-KI-Verordnung (EU AI Act) ist veröffentlicht. Viele Hersteller von Medizinprodukten und IVD sowie andere Akteure im Gesundheitswesen stehen vor der großen Aufgabe, den über 140 Seiten umfassenden Gesetzestext zu verstehen und die Anforderungen zu erfüllen. Beachten Sie: Verstöße gegen den AI Act werden mit Geldstrafen in Höhe von bis zu 7 % des jährlichen Umsatzes geahndet.
Dieser Artikel erspart Recherchearbeit und gibt konkrete Tipps, wie Hersteller (und andere Akteure) die Anforderungen schnell erfüllen.
Nutzen Sie das kostenlose AI Act Starter-Kit, um schnell Konformität zu erreichen und im nächsten QM-Audit zu glänzen.
Medizinprodukte- und IVD-Hersteller ohne KI-basierte Produkte sollten diesen Artikel lesen.
1. Wen der AI Act betrifft (nicht nur Hersteller!)
Die EU-KI-Verordnung 2024/1689 (kurz: AI Act) betrifft viele Akteure im Gesundheitswesen. Der AI Act ist meist anwendbar, wenn die dafür hergestellten oder dort verwendeten Produkte ein „KI-System“ sind oder beinhalten. Er betrifft auch die Hersteller, welche KI anwenden, beispielsweise LLMs bei der Entwicklung von Medizinprodukten und bei regulatorischen Prozessen.
Leider definiert der AI Act den Begriff „KI-System“ sehr schwammig.
Maschinengestütztes System, das für einen in unterschiedlichem Grade autonomen Betrieb ausgelegt ist und das nach seiner Betriebsaufnahme anpassungsfähig sein kann und das aus den erhaltenen Eingaben für explizite oder implizite Ziele ableitet, z. B. Ausgaben wie etwa Vorhersagen, Inhalte, Empfehlungen oder Entscheidungen erstellt werden, die physische oder virtuelle Umgebungen beeinflussen können;
KI-Verordnung, Artikel 3 (1)
Eine Kritik dieser Definition findet sich in diesem Fachartikel.
Eine hilfreiche Vereinfachung der Definition besteht darin, die KI-Systeme im Sinne der KI-Verordnung mit Software-Systemen oder Software-Komponenten gleichzusetzen, die Verfahren des maschinellen Lernens (ML) nutzen, d. h. ein „ML-Modell“ sind oder beinhalten.
Der AI Act ist eines von vielen Gesetzen, die Hersteller KI-basierter Medizinprodukte beachten müssen. Sie finden hier eine Übersicht der regulatorischen Anforderungen an diese KI-basierten Produkte und hier eine Übersicht zum Einsatz der KI im Gesundheitswesen.
1.1 Anbieter/Hersteller
1.1.1 Definitionen
Alleine die Tatsache, dass ein IVD oder ein Medizinprodukt Verfahren des maschinellen Lernens verwendet, bedeutet nicht zwingend, dass diese Produkte und deren Hersteller nennenswerte Anforderungen des AI Act (Anforderungen an Hochrisiko-Systeme) erfüllen müssen.
Das ist nur der Fall, falls zwei Bedingungen erfüllt sind (s. Abb. 1):
- Das Produkt (Medizinprodukt bzw. IVD) fällt in eine Klasse, bei der eine Benannte Stelle in das Konformitätsbewertungsverfahren nach MDR bzw. IVDR eingebunden werden muss. Das sind die Medizinprodukte der Klassen I*, IIa, IIb und III bzw. IVD der Klassen As, B, C und D.
- Die KI (präziser: das Machine-Learning-Modell) stellt entweder selbst das Produkt dar oder dient als „Sicherheitsbauteil“ eines Produkts.
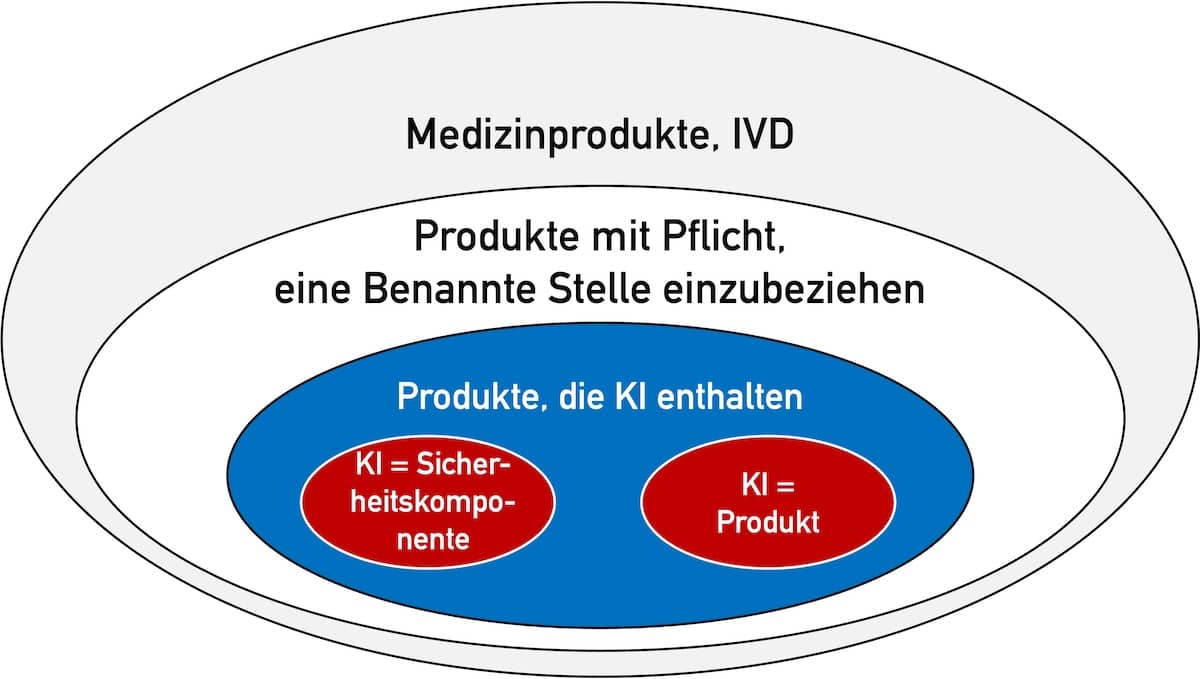
Der AI Act definiert den Begriff „Sicherheitsbauteil“.
Bestandteil eines Produkts oder KI-Systems, der eine Sicherheitsfunktion für dieses Produkt oder KI-System erfüllt oder dessen Ausfall oder Störung die Gesundheit und Sicherheit von Personen oder Eigentum gefährdet;
KI-Verordnung, Artikel 3 (14)
Somit kann ein Sicherheitsbauteil sowohl ein Teil einer Standalone-Software als auch Teil eines physischen Produkts sein.
Produkte der niedrigsten Klasse (z. B. MDR Klasse I), welche Verfahren des maschinellen Lernens verwenden, fallen zwar unter die KI-Verordnung, aber sie zählen nicht als Hochrisiko-Produkte. Daher müssen sie in der Regel keine in der Praxis relevanten (geprüften) Anforderungen erfüllen.
1.1.2 Künftige Ergänzung durch
„Only certain AI systems are subject to regulatory obligations and oversight under the AI Act” dürfte für viele der Lieblingssatz aus dem Entwurf der Leitlinie „on the definition of an artificial intelligence system established by Regulation (EU) 2024/1689 (AI Act)” lauten. Denn er stellt klar, dass nicht alle KI-basierten Medizinprodukte bzw. IVD unter den AI Act fallen.
Damit klärt er aber weder, was KI-basierte Produkte sind, noch, welche dieser Produkte vom AI Act betroffen sind.
Auf 13 Seiten mühen sich die Autoren um Klarstellung. Das gelingt nicht immer. So heißt es etwa an einer Stelle, dass KI zu unterscheiden sei von „systems that are based on the rules defined solely by natural person“. An anderer Stelle schreiben die Autoren, dass KI-Systeme Ziele verfolgen müssten. Dazu zählten „clearly stated goals that are directly encoded by the developer into the system“.
Was zumindest klarer wird, ist, dass die Definition von KI sich nicht auf das Machine Learning beschränkt, sondern auch „reasoning“ und „logical inference“ einbezieht, wie das bei „early generation expert systems“ der Fall sei.
Dass bereits „operations such as sorting, searching, matching, chaining“ dazuzählen können, wundert. Denn welche Software führt diese Operationen nicht aus? Hingegen zählen die Autoren die logistische Regression nicht mehr zur KI, weil diese nicht über „basic data processing“ hinausginge.
Es wird nur wenige Medizinprodukte geben, die maschinelles Lernen (ML) verwenden und nicht als Hochrisiko-Produkte zählen. Denn KI/ML-basierte Produkte enthalten immer Software oder sind eigenständige Software. Für derartige Medizinprodukte greift die Regel 11 der MDR. Diese wird inzwischen (leider) so ausgelegt, dass insbesondere Software as a Medical Device mindestens als Klasse IIa klassifiziert werden muss.
Für IVD sind die Klassifizierungsregeln nach Anhang VIII der IVDR anzuwenden.
Eine praktikable Vereinfachung ist es daher, ML-basierte IVD und Medizinprodukte zunächst mit Hochrisiko-Produkten gleichzusetzen. Beispiele für Ausnahmen von dieser Regel nennt Kapitel 1.1.2.
1.1.3 Beispiele für Hochrisiko-Produkte und Nicht-Hochrisiko-Produkte
| Kategorie: KI-System ist … | Beispiel | Einstufung |
| das Medizinprodukt selbst | KI-/ML-basierte Standalone-Software, die dazu dient, maligne Hautveränderungen zu diagnostizieren (Je nach Architektur und Funktionalität der Software bildet das KI-System für die Diagnose nur einen Teil des Produkts. Damit wäre dieses „Diagnose-Modul“ als Sicherheitsbauteil zu betrachten.) | Hochrisiko-Produkt |
| Teil eines Sicherheitsbauteils des Medizinprodukts | KI/ML-basierte Software einer Dialysemaschine, welche die Ultrafiltration steuert | Hochrisiko-Produkt |
| Teil eines Bauteils des Produkts, das kein Sicherheitsbauteil ist | KI/ML-basierte Softwarekomponente, welche die Nutzung des Produkts auswertet, um dem Hersteller Hinweise über den Verstoß gegen Lizenzbedingungen zu geben | Kein Hochrisiko-Produkt |
| eine General-Purpose-AI (konkret ein LLM eines der großen Anbieter, etwa OpenAI) | LLM zum Schreiben von Software-Code, der Teil eines Medizinprodukts wird | Kein Hochrisiko-Produkt |
1.1.4 Sonderfall „Bestimmte KI-Systeme“
In Artikel 50 führt der AI Act den Begriff der „bestimmten KI-Systeme“ ein, ohne ihn zu definieren. Unter Umständen fallen auch IVD und Medizinprodukte darunter (s. Tabelle 2).
| Eigenschaft des Produkts | Beispiel | Konsequenz |
| KI-System ist für direkte Interaktion mit natürlichen Personen bestimmt | Software zur Diagnose maligner Hautveränderungen | Die Anbieter (Hersteller) müssen die Personen über den Einsatz der KI informieren. |
| KI-System dient der Emotionserkennung | Software für den Einsatz in der Psychotherapie | Betreiber müssen die betroffenen Personen über den Einsatz der KI und die Verwendung der Daten informieren. |
Auf diese „bestimmten KI-Systeme“ geht dieser Beitrag im Folgenden nicht mehr ein, weil die Anforderungen durch die MDR bzw. IVDR im Wesentlichen bereits abgedeckt sind.
1.2 Sonstige Akteure
Die KI-Verordnung stellt bei Hochrisiko-Produkten (und damit IVD und Medizinprodukten) Anforderungen nicht nur an die Hersteller, sondern auch an weitere Akteure:
- Betreiber (z. B. Krankenhäuser, Labore)
- Importeure/Einführer
- Händler
Die Anforderungen an die Betreiber beschreibt Kapitel 2.3 sowie noch ausführlicher dieser Artikel.
2. Diese Anforderungen stellt der AI Act
2.1 Übersicht
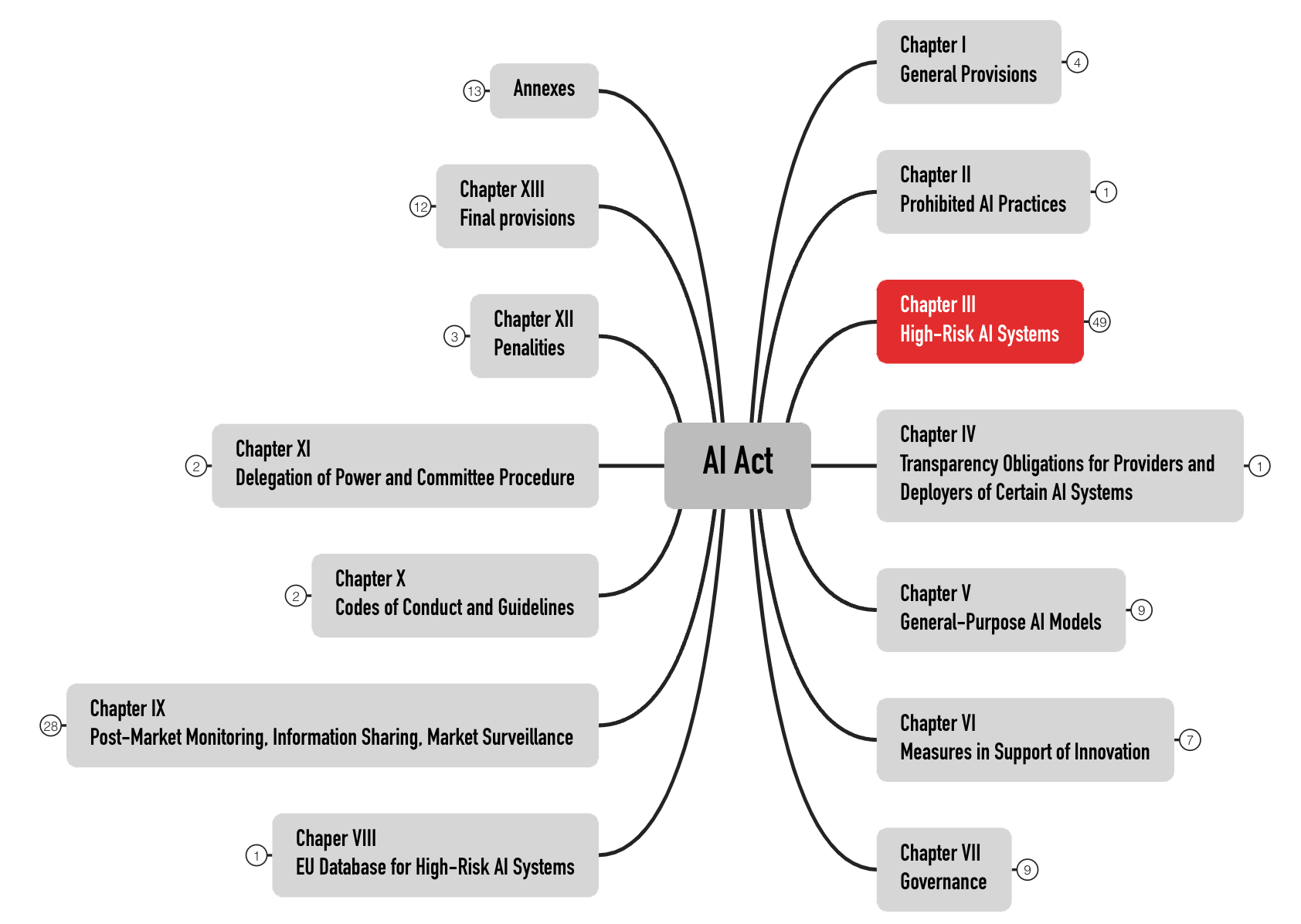
Der AI Act formuliert die Anforderungen an die Hochrisiko-Produkte in Kapitel III. Mit 49 Artikeln ist es das umfangreichste Kapitel (s. Abb. 2). Produkte anderer Risikokategorien behandelt dieser Beitrag nicht.
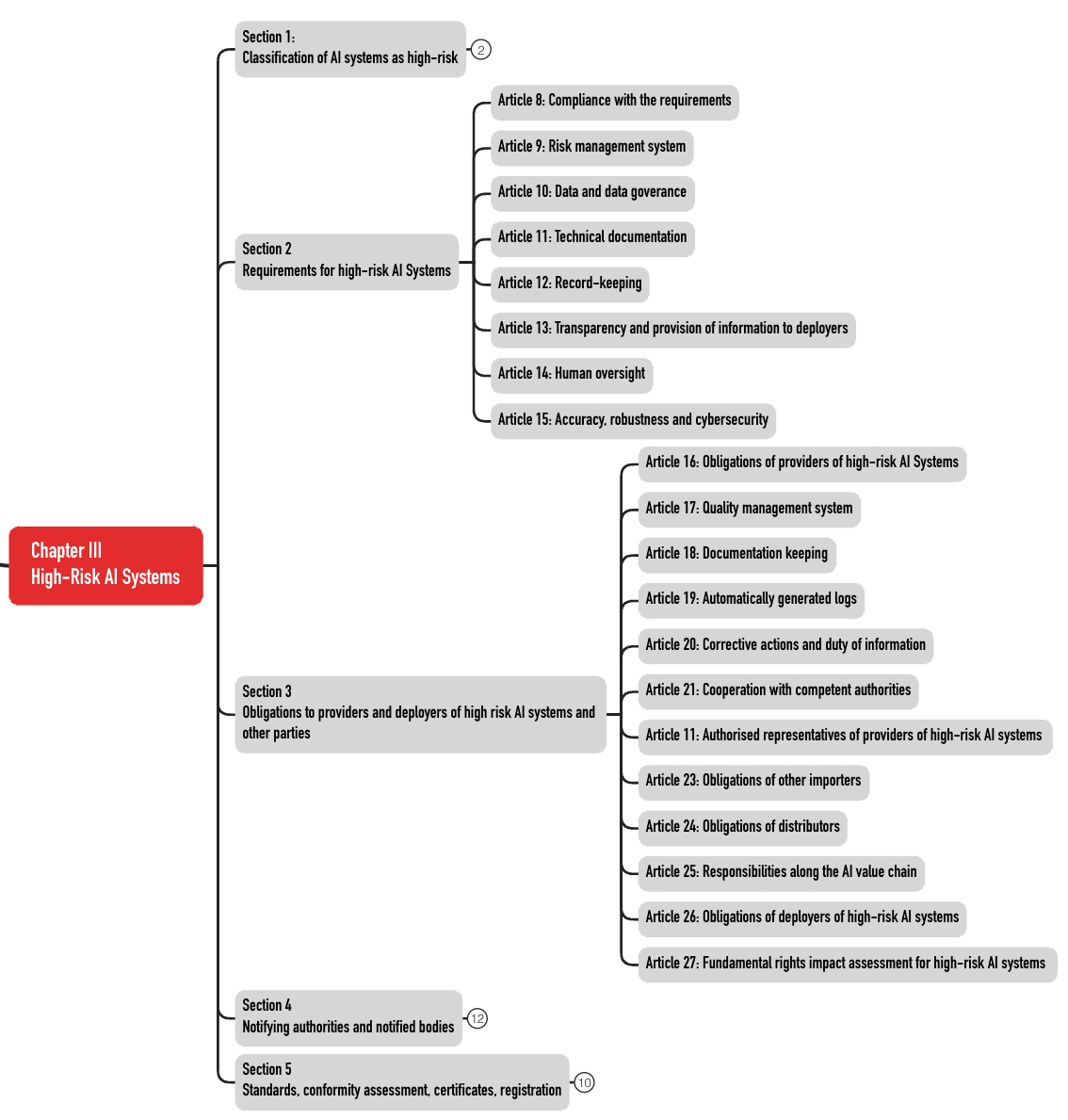
Diese 49 Artikel sind in fünf Abschnitte (Sections) aufgeteilt (s. Abb. 3). Die Anforderungen an die KI-Systeme finden sich in den Abschnitten 2 und 3.
Laden Sie sich das AI Act Starter-Kit herunter. Es enthält nicht nur die vollständigen Mindmaps als PDF und im XMind-Format, sondern auch weitere hilfreiche Informationen.
2.2 Anforderungen an die Hersteller
Dieser Abschnitt fasst die wichtigsten Anforderungen des AI Act an die Hersteller zusammen.
| Anforderung | Artikel | Kommentar |
| Risikomanagementsystem | 9 | Die Schutzziele betreffen auch die Grundrechte. Das Testen wird explizit gefordert. Konkrete Risiken, die die Hersteller betrachten müssen, werden vorgegeben, z. B. „data poisoning“ und Cybersecurity-Probleme. |
| Daten-Governance | 10 | Vorgaben zum Umgang mit Trainings-, Validierungs- und Testdaten |
| Technische Dokumentation (TD) | 11 | Die TD kann und soll mit der TD von Medizinprodukten/IVD zusammengelegt werden. |
| Aufzeichnungspflichten | 12 | Gemeint ist eine Protokollierung u. a. der Nutzung und Input-Daten, beispielsweise im Sinne von Audit-Trails, um die Post-Market Surveillance zu vereinfachen. |
| Informationen | 13 | Es wird eine elektronische(!) Gebrauchsanweisung gefordert. |
| Menschliche Aufsicht | 14 | Die menschliche Aufsicht bei der Nutzung der Systeme soll helfen, Risiken zu vermindern. Es gibt viele Querbezüge zur Gebrauchstauglichkeit dieser Produkte. |
| Genauigkeit, Robustheit und Cybersicherheit | 15 | Diese Anforderungen betreffen nicht nur die Entwicklung der Produkte, sondern auch organisatorische Maßnahmen. Der AI Act schließt selbstlernende Systeme nicht aus, im Gegensatz zu einigen deutschen Benannten Stellen bei Medizinprodukten. |
| Qualitätsmanagementsystem | 16, 17 | Dieses System kann und sollte mit dem bestehenden QMS betrieben werden. Es setzt zusätzliche/geänderte Vorgabedokumente voraus (s. u.). |
| Konformitätsbewertungsverfahren, Konformitätserklärung und CE-Kennzeichnung | 16, 43, 47, 48 | Es sind Benannte Stellen einzubeziehen (das werden hoffentlich die gleichen Benannten Stellen sein wie die für die Medizinprodukte/IVD). |
| Pflicht zur Registrierung in einer Datenbank (bei Medizinprodukten und IVD nicht notwendig!) | 16, 49 | Vergleichbar mit EUDAMED |
| Vigilanzsystem, Zusammenarbeit mit Behörden | 16, 20, 21 | Vergleichbar der Vigilanz bei MDR und IVDR |
| Anforderungen an Barrierefreiheit | 16 | |
| Bestellung eines EU-Bevollmächtigten | Falls der Hersteller („Anbieter“) nicht in der EU sitzt |
2.3 Anforderungen an die Betreiber
2.3.1 Was ein Betreiber ist
Die KI-Verordnung definiert den Begriff Betreiber:
Eine natürliche oder juristische Person, Behörde, Einrichtung oder sonstige Stelle, die ein KI-System in eigener Verantwortung verwendet, es sei denn, das KI-System wird im Rahmen einer persönlichen und nicht beruflichen Tätigkeit verwendet;
KI-Verordnung, Artikel 3 (4)
Damit zählt ein Medizinproduktehersteller, der LLMs verwendet (z. B. Microsoft Copilot) als Betreiber.
Im Gegensatz zu MDR und IVDR kennt die KI-Verordnung das Konzept der Eigenherstellung bzw. der Inhouse-IVD nicht. Aber wenn eine Gesundheitseinrichtung ein KI-System selbst entwickelt und in Betrieb nimmt, zählt sie der AI Act zu den Anbietern. Denn definitionsgemäß gelten auch diejenigen Betreiber als Anbieter, die KI-Systeme „unter ihrem eigenen Namen […] in Betrieb nehmen“.
Eine natürliche oder juristische Person, Behörde, Einrichtung oder sonstige Stelle, die ein KI-System oder ein KI-Modell mit allgemeinem Verwendungszweck entwickelt oder entwickeln lässt und es unter ihrem eigenen Namen oder ihrer Handelsmarke in Verkehr bringt oder das KI-System unter ihrem eigenen Namen oder ihrer Handelsmarke in Betrieb nimmt, sei es entgeltlich oder unentgeltlich;
KI-Verordnung, Artikel 3 (3)
Beispielsweise zählt ein Krankenhaus oder medizinisches Labor, das ein KI-System selbst entwickelt und z. B. für diagnostische Zwecke in Betrieb nimmt, als Anbieter.
Die KI-Verordnung gilt explizit auch für „Produkthersteller, die KI-Systeme zusammen mit ihrem Produkt unter ihrem eigenen Namen oder ihrer Handelsmarke in Verkehr bringen oder in Betrieb nehmen“ (Artikel 2(1)e)).
MDR und IVDR verbieten Eigenherstellungen von Produkten, die es in ähnlicher Version kommerziell und CE-gekennzeichnet am Markt gibt, unabhängig davon, ob diese ein KI-System enthalten oder nicht.
Allerdings gelten für die Vorgaben zur Eigenherstellung Übergangsfristen. Für Inhouse-IVD findet Artikel 5(5)d der IVDR erst ab dem 31. Dezember 2030 Anwendung.
2.3.2 Was die Pflichten der Betreiber sind
Die Betreiberpflichten formuliert der AI Act v. a. in Artikel 26.
| Pflicht | Kommentar |
| Zweckbestimmungsgemäßen Gebrauch sicherstellen | Sicherstellen, dass die Anwender über die notwendigen Kompetenzen verfügen und das Produkt gemäß der Gebrauchsanweisung nutzen. Das schließt ein, dass die Input-Daten der Zweckbestimmung genügen. Der AI Act führt nicht aus, weshalb diese Daten auch „repräsentativ“ sein sollen. Ist die Annahme, dass die Anwender das System trainieren? |
| Vigilanz | Wenn die Betreiber vermuten, dass das System nicht sicher ist, müssen sie den Anbieter und(!) die Marktaufsichtsbehörden informieren. |
| Protokolle aufbewahren | Die KI-Systeme müssen Protokolle für die Post-Market Surveillance erzeugen (z. B. Audit-Logs). Diese müssen die Betreiber aufbewahren. Dass sie dabei in den Konflikt mit dem Datenschutz geraten können, ist den Autoren des AI Act bewusst. |
| Über den Einsatz informieren | Die Betreiber müssen die Arbeitnehmenden und deren Vertreter (z. B. den Betriebsrat) informieren. |
| Datenschutz gewährleisten | Der AI Act verlangt von den Betreibern, ihren Pflichten durch die DSGVO nachzukommen. |
Bitte beachten Sie den Artikel zu den Anforderungen des AI Act an die Betreiber (Deployer).
3. Ab wann die Anforderungen gelten
Die Zeitpunkte für das Inkrafttreten und den Geltungsbeginn definiert der Artikel 113.
| Datum | Aktion |
| 12.07.2024 | EU verabschiedet KI-Verordnung |
| 01.08.2024 | KI-Verordnung tritt in Kraft |
| 02.02.2025 | Verbot der „verbotenen Praktiken“ wird wirksam |
| 02.08.2025 | Vorschriften für KI-Modelle mit allgemeinem Verwendungszweck werden wirksam (wenn diese ab jetzt in den Verkehr gebracht werden) |
| 02.08.2026 | Die KI-Verordnung gilt weitgehend, auch für viele Hochrisiko-Produkte |
| 02.08.2027 | Ab jetzt zählen Produkte gemäß Artikel 6(1) (das sind IVD und Medizinprodukte sowie Sicherheitskomponenten) als Hochrisiko-Produkte. Das dürfte für IVD- und Medizinproduktehersteller die wichtigste Frist sein. |
| 02.08.2027 | Vorschriften für KI-Modelle mit allgemeinem Verwendungszweck werden wirksam, wenn diese vor dem 01.08.2025 in den Verkehr gebracht wurden. |
4. Das sollten Hersteller jetzt tun
Die Entwicklung von IVD und Medizinprodukten dauert meist Jahre. Daher sind die Hersteller gut beraten, sich ab sofort mit den Anforderungen des AI Act auseinanderzusetzen und nicht bis zum Ende der Übergangsfrist zu warten.
Zudem überprüfen Benannte Stellen bei Audits gezielt, ob die Hersteller Änderungen an den regulatorischen Anforderungen systematisch identifizieren und notwendige Maßnahmen ergreifen. Die KI-Verordnung ist ein Beispiel für solch eine regulatorische Änderung.
Die Direktorin „Global Regulatory Affairs“, Carmen Bellebna, berichtet in dieser Episode darüber, wie sie und ihr Unternehmen DeepEye die Anforderungen des AI Act erfüllen.
Diese und weitere Podcast-Episoden finden Sie auch hier.
4.1 Kompetenz aufbauen
Die Hoffnung, dass gute Data Scientists in den eigenen Reihen ausreichen, um die gesetzlichen Anforderungen nach Kompetenz zu erfüllen, dürfte nicht erfüllt werden. Umfassenderes Wissen ist notwendig, beispielsweise bei den folgenden Aktivitäten und Bereichen:
- Validierung der Systeme zum Sammeln und Verarbeiten (Pre-Processing) der Trainings-, Validierungs- und Testdaten
- Risikomanagement, das neue Schutzziele und neue Risiken einbeziehen muss
- Umsetzung künftig erscheinender einschlägiger (harmonisierter) Normen
- Testmethoden für KI-Modelle
- Methoden für die Interpretability (= Explainability + Transparency) dieser Modelle
- Datenschutz bei KI-Modellen (insbesondere bei Trainings-, Validierungs- und Testdatensätzen sowie bei den Protokollen)
Die ISO 13485 verpflichtet die „Organisationen“ sogar dazu, die Kompetenzen spezifisch für jedes einzelne Entwicklungsprojekt/Produkt festzulegen.
Der AI Act verlangt in Artikel 4, die Kompetenz „nach besten Kräften“ sicherzustellen. Es wird nicht ausgeführt, welche Kompetenzen gemeint sind. Das KI-Gesetz der EU schließt auch die Betreiber und Anwender von seinen Forderungen nicht aus.
Der KI-Leitfaden des Johner Instituts, der auch von den Benannten Stellen genutzt wird, enthält wichtige Checklisten. Anhand dieser können Sie die Konformität Ihrer KI-basierten Medizinprodukte prüfen und herstellen.
4.2 QM-Vorgaben erweitern
Die Hersteller werden ihre Vorgabedokumente wie Verfahren- und Arbeitsanweisungen, Checklisten und Templates anpassen und ergänzen, um die zusätzlichen Anforderungen der KI-Verordnung zu erfüllen.
Diese Vorgabedokumente müssen fortlaufend darauf überprüft werden, ob sie dem Stand der Technik (insbesondere harmonisierten Normen) entsprechen, und geschult werden.
4.3 Externe Partner auswählen und managen
Der AI Act wirkt sich nicht nur auf das Arbeiten innerhalb der Unternehmen aus, sondern auch auf deren Interaktion mit Externen. Das betrifft beispielsweise die folgenden Tätigkeiten:
- Lieferanten identifizieren, qualifizieren und fortlaufend bewerten (z. B. Open Source Modelle, Entwicklungsdienstleister, Benannte Stellen)
- Händler auswählen, instruieren, überprüfen
- Ggf. EU-Bevollmächtigte auswählen
- Partner für Schulungen und Beratung auswählen (Tipp: Johner Institut 🙂 )
- KI-Reallabore finden
- Förderprogramme recherchieren, Forschungszulagen beantragen
5. Hintergrund, Kritik, Aktuelles
a) Hintergrund
Auf ihrer Webseite Neue Vorschriften für künstliche Intelligenz – Fragen und Antworten kündigt die EU an, KI in allen Branchen risikobasiert regulieren und die Einhaltung der Vorschriften überwachen (lassen) zu wollen.
Die EU sieht die Chancen des Einsatzes von KI, aber auch deren Risiken. Sie erwähnt explizit die Gesundheitsversorgung und möchte durch eine EU-weite Regulierung die Fragmentierung des Binnenmarkts verhindern.
Die Regulierung soll sowohl Hersteller als auch Anwender betreffen. Die umzusetzenden Vorschriften hängen davon ab, ob das KI-System als Hochrisiko-KI-System eingestuft wird oder nicht. Produkte, die unter die MDR oder IVDR fallen und für die die Beteiligung einer Benannten Stelle gefordert ist, gelten als Hochrisikoprodukte.
Ein Schwerpunkt der Regulierung wird die biometrische Fernidentifizierung betreffen.
Die Regulierung sieht auch eine Überwachung und Auditierung vor; zudem erstreckt sie sich auf importierte Produkte. Die EU plant ein Zusammenspiel dieser Regulierung mit der neuen Maschinenverordnung (wohlgemerkt: Verordnung), die die bisherige Maschinenrichtlinie ablösen würde.
Es soll ein spezieller Ausschuss gegründet werden: „Der Europäische Ausschuss für künstliche Intelligenz soll sich aus hochrangigen Vertretern der zuständigen nationalen Aufsichtsbehörden, des Europäischen Datenschutzbeauftragten und der Kommission zusammensetzen.“
b) Kritik
Wie immer betont die EU, dass der Standort gefördert werden soll und KMU nicht über Gebühr belastet werden dürfen. Diese Aussagen finden sich auch in der MDR. Aber keine der Verordnungen scheint diesem Anspruch gerecht zu werden.
Kritik an Definitionen
Die Definition ist unscharf. Zu den Verfahren der künstlichen Intelligenz zählen nicht nur das maschinelle Lernen, sondern auch:
– Logik- und wissensgestützte Konzepte, einschließlich Wissensrepräsentation, induktiver (logischer) Programmierung, Wissensgrundlagen, Inferenz- und Deduktionsmaschinen, (symbolischer) Schlussfolgerungs- und Expertensysteme;
– Statistische Ansätze, Bayessche Schätz-, Such- und Optimierungsmethoden
Dieser breite Anwendungsbereich kann dazu führen, dass viele Medizinprodukte, die Software enthalten, in den Anwendungsbereich der KI-Verordnung fallen. Jeder in Software gegossene Entscheidungsbaum wäre demnach ein KI-System.
Die Verordnung definiert zwar den Begriff „Sicherheitskomponente“, aber sie verwendet dafür den nicht definierten Begriff einer Sicherheitsfunktion.
Bestandteil eines Produkts oder KI-Systems, der eine Sicherheitsfunktion für dieses Produkt oder KI-System erfüllt oder dessen Ausfall oder Störung die Gesundheit und Sicherheit von Personen oder Eigentum gefährdet;
AI Act, Artikel 3 (14)
Andere Begriffe definiert die KI-Verordnung nicht in Übereinstimmung mit der MDR, z. B. „Post-Market Monitoring“ oder „serious inicident“.
Kritik am Anwendungsbereich
Die KI-Verordnung wendet sich explizit an Medizinprodukte und IVD. Es gibt damit eine Dopplung an Anforderungen. MDR und IVDR fordern bereits Cybersecurity, ein Risikomanagement, die Post-Market Surveillance, ein Meldesystem, eine Technische Dokumentation, ein QM-System usw. Hersteller müssen nun die Konformität mit zwei Verordnungen nachweisen!
Kritik an risikobasiertem Ansatz
Die Verordnung gilt weitgehend unabhängig davon, wofür die KI im Medizinprodukt eingesetzt wird. Eine Gefährdung der Gesundheit, unabhängig von der Höhe des Risikos, macht das Produkt zum Hochrisikoprodukt.
Selbst eine KI, mit der ein verschleißärmerer Betrieb eines Motors realisiert werden soll, würde unter die EU-Verordnung fallen. Als Konsequenz werden Hersteller sich genau überlegen, ob sie von KI-Verfahren Gebrauch machen. Das kann sich nachteilig auf die Innovation, aber auch auf die Sicherheit und Leistungsfähigkeit von Produkten auswirken. Denn i. d. R. setzen die Hersteller KI ein, um die Sicherheit, Leistungsfähigkeit und Wirksamkeit von Produkten zu verbessern. Andernfalls dürften sie KI gar nicht verwenden.
Mit dieser Forderung ist der Einsatz von KI in Situationen erschwert, in denen Menschen nicht mehr ausreichend schnell reagieren können. Dabei könnte genau in diesen Situationen der Einsatz von KI besonders hilfreich sein.
Wenn wir neben jedes Gerät eine Person setzen müssen, die den Einsatz der KI „wirksam beaufsichtigt“, bedeutet dies das Ende der meisten KI-basierten Produkte.
Kritik an der Klassifizierung
Die unglückliche Regel 11 teilt Software – unabhängig vom Risiko – in den allermeisten Fällen in die Klassen II a oder höher ein. Damit gelten für Medizinprodukte die umfangreichen Anforderungen an Hochrisikoprodukte. Die negativen Auswirkungen dieser Regel 11 werden durch die KI-Verordnung verstärkt.
Kritik an der Realitätsnähe der Anforderungen
In Artikel 10 fordert die KI-Verordnung:
„training, validation, and testing data sets shall be relevant, representative, free of errors and complete.“
Real-World-Daten sind selten fehlerfrei und vollständig. Es bleibt auch unklar, was mit „complete“ gemeint ist. Müssen alle Datensätze vorliegen (was auch immer das bedeutet) oder alle Daten eines Datensatzes?
Kritik an der Übergriffigkeit der Verordnung
Der Anhang der KI-Verordnung verlangt von den Herstellern, den Behörden einen vollständigen Remote-Zugriff auf die Trainings-, Validierungs- und Testdaten zu verschaffen, sogar durch eine API:
Die technische Dokumentation wird von der notifizierten Stelle geprüft. Sofern es relevant ist und beschränkt auf das zur Wahrnehmung ihrer Aufgaben erforderliche Maß erhält die notifizierte Stelle uneingeschränkten Zugang zu den verwendeten Trainings-, Validierungs- und Testdatensätzen, gegebenenfalls und unter Einhaltung von Sicherheitsvorkehrungen auch über API oder sonstige einschlägige technische Mittel und Instrumente, die den Fernzugriff ermöglichen.
AI Act, Annex IV, Abschnitt 4
Vertrauliche Patientendaten über einen Remote-Zugriff zugänglich zu machen, steht im Konflikt mit der gesetzlichen Forderung nach Data Protection by Design. Gesundheitsdaten zählen zur besonders schützenswerten Kategorie personenbezogener Daten.
Eine externe API zu den Trainingsdaten zu entwickeln und bereitzustellen, bedeutet für die Hersteller einen zusätzlichen Aufwand.
Dass Behörden mit diesem Zugriff die Daten bzw. die KI mit vertretbarem Aufwand und in vertretbarer Zeit herunterladen, analysieren und bewerten können, ist unrealistisch.
Bei anderen, oft sogar kritischeren Daten und Informationen zum Produktdesign und zur Produktion (z. B. Source-Code oder CAD-Zeichnungen) würde niemand ernsthaft verlangen, dass die Hersteller den Behörden einen Remote-Zugriff gewähren müssen.
c) Aktuelles
Stand Oktober 2025 sind noch keine Organisationen für den AI Act benannt. Das heißt, dass es noch keine Benannte Stellen für den AI Act gibt. Es ist jedoch zu erwarten, dass alle für die MDR bzw. IVDR Benannten Stellen durch eine Scope-Erweiterung auch für den AI Act zuständig werden.
Ferner soll bei Medizinprodukten und IVD die ZLG die „Competent Authority“ bleiben (auch für den AI Act) und nicht etwa die Bundesnetzagentur.
Die Akkreditierung der Zertifizierstellen für die Norm ISO 42001 erfolgt davon unabhängig (durch die DAkkS).
Die Norm ist derzeit weder für die MDR und IVDR noch für den AI Act harmonisiert.
Lassen Sie sich nur von Organisationen zertifizieren, die dafür akkreditiert sind. Andernfalls ist das Zertifikat wertlos.
6. Fazit und Zusammenfassung
Der AI Act ist da und wird bleiben. Daran ändert auch die breite, teilweise vernichtende Kritik an dieser EU-KI-Verordnung nichts, die wir an anderer Stelle zusammenfassen.
a) Jetzt starten, aber risikobasiert
Daher müssen sich Hersteller mit der Verordnung auseinandersetzen.
Tipp: Starten Sie zeitnah, denn einige Vorgaben lassen sich bei einem bereits entwickelten Produkt nachträglich nur noch schwer erfüllen.
Dabei sollten Sie risikobasiert vorgehen:
- Wie bei der MDR und IVDR hilft es, einerseits frühzeitig zu starten, um Risiken in Audits und für die plangemäße („Time & Budget“) Entwicklung und Vermarktung der Produkte zu minimieren.
- Andererseits müssen sich die Hersteller bewusst sein, dass die Normen für den AI Act erst noch geschrieben und harmonisiert werden. Das kann das Nacharbeiten der QM-Dokumente erfordern.
Der Termin, an dem Medizinprodukte und IVD die Anforderungen an Hochrisiko-Produkte erfüllen müssen, steht fest. Unabhängig von der (schleppenden) Harmonisierung von Normen.
b) Ein „Change Project“ initiieren
Der AI Act bedeutet mehr als „nur ein weiteres Gesetz“. Er betrifft viele Abteilungen:
- Forschung und Entwicklung inkl. Usability Engineering und Data Science
- Qualitätsmanagement inkl. Lieferanten-Management
- Regulatory Affairs inkl. Vigilanz und Auswahl (neuer?) Benannter Stellen
- Datenschutz und IT Security
- Risikomanagement
- Produktmanagement und Customer-Support
Diese Änderungen erfordern Ressourcen (Geld, ausgebildete Personen) und eine abteilungsübergreifende Planung. Daher sollten die verantwortlichen Führungskräfte ein Change-Projekt aufsetzen und nicht nur Regulatory Affairs mit einem „Mach mal“ beauftragen.
Das AI Act Starter-Kit verschafft einen ersten Überblick über die anstehenden Aufgaben.
Die Experts des Johner Instituts helfen Herstellern in den Bereichen AI, QM und RA, mit einem schlanken Projekt die Anforderungen des AI Act Schritt für Schritt zu erfüllen.
Änderungshistorie
- 2025-10-20: Im Kapitel 5.b) Unterüberschrift „Kritik an der Übergriffigkeit der Verordnung“ die Referenz auf den Anhang IV sowie das passende Zitat eingefügt. Im Entwurf des AI Acts fand sich diese Forderung im Artikel 64.
- 2025-10-18: In Kapitel 2.2 klargestellt, dass die Registrierungspflichten Medizinproduktehersteller nicht betreffen
- 2025-10-17: Aussagen in Kapitel 5 c) zur Akkreditierung präzisiert und Warnung vor nicht akkreditierten Zertifizierstellen eingefügt
- 2025-10-09: Mehrere Änderungen
- Hinweise zum Scope im ersten Absatz im Abschnitt 1 ergänzt
- Beispiel im Abschnitt 1.1.3 hinzugefügt
- In 2.3.1 nach Definitionsbox Beispiel ergänzt
- Kapitel 5 c) („Aktuelles“) eingefügt
- 2025-05-05: Hinweis auf Podcast hinzugefügt
- 2025-04-15: Hinweis am Anfang auf Artikel für Hersteller ohne KI-basierte Produkte ergänzt
- 2025-02-22: Kapitel 5 neu (aus Artikel zum Machine Learning übernommen, umstrukturiert und aktualisiert). Abschnitt 1.1.2 eingefügt. Folgende Nummerierung angepasst
- 2024-10-22: Erste Version des Artikels veröffentlicht


Vielen Dank für die griffige Zusammenfassung. Beim Lesen des Artikels ist mir aufgefallen, das hier wie auch im Gesetzestext der Begriff „Validierung“ nicht eindeutig verwendet wird. in Kap. 4.1. wird auf das Sammeln von Trainings-, Validierungs- und Testdaten hingewiesen (also auf den „training – validation -test“-Zyklus im Rahmen der AI-Entwicklung), der Link jedoch verweist auf „Validierung der Systeme“, was im Kontext von Medizinprodukten eine ganz andere Bedeutung hat. Über eine Präzisierung der Begrifflichkeiten würde ich mich freuen, sowie über eine Einschätzung Ihrerseits, wie die Verwendung des Begriffs „Validierung“ im EU AI Act aus Ihrer Sicht zu lesen ist.
Die FDA verfolgt hier eine konsistente Linie und verwendet in ihren Veröffentlichungen (u.a. auch in dem von Ihnen verlinkten Glossar) konsequent „Tuning“, wenn es um den AI-Entwicklungzyklus geht („trainig – tuning – test“). Der Begriff „Validierung“ bleibt der „System-Validierung“ vorbehalten.
Sehr geehrter Herr Zeltner,
danke für Ihre wertschätzende Rückmeldung und Ihren sehr wertvollen Hinweis! Ich stimme Ihnen absolut zu, dass wir wieder das Thema einer mangelnden Definition des Begriffs „Validierung“ haben. Ich stimme Ihnen auch zu, dass die FDA etwas weiter ist, wobei der FDA Validation Guidance auch eine andere Interpretation zuließe.
Zumindest definiert der AI-Act den Begriff „Validierungsdaten“:
Weil der Begriff mit einer Ausnahme nur im Kontext von Trainings-, Validierungs- und Testdaten (in dieser Reihenfolge) verwendet wird, können wir davon ausgehen, dass die EU im AI Act unter Validierung nicht(!) das versteht, was wir „Medizinprodukte-Menschen“ darunter verstehen, nämlich eine Prüfung, ob das Produkt geeignet ist, die Zweckbestimmung und die Stakeholder-Anforderungen (z.B. fachlichen Anforderungen) zu erfüllen.
Leider gibt es nicht einmal bei den KI-Experten immer Einigkeit, was die Validierungen sind, weil manche von Trainings-, Test- und Validierungsdaten sprechen.
Ausführlichere Gedanken finden Sie im Artikel zur Verifizierung und Validierung und in dem dort im Abschnitt 4.a) verlinkten Paper.
Nochmals besten Dank!
Herzliche Grüße, Christian Johner
Sehr geehrter Herr Johner,
vielen Dank für diesen sehr guten Artikel. Ich hätte eine Frage zum Scope des AI Acts. Betrifft der AI ACt nur Produkte, die nach Ihrer Inbetriebname weiter lernen? Oder sind auch Produkte betroffen die am Ende der Entwicklund „eingefroren“ werden und während des Betriebs nicht weiter lernen?
Herzlichen Dank und viele Grüße
Maria Pinkerneil
Sehr geehrte Frau Pinkerneil,
der AI Act unterscheidet kaum zwischen weiterlernenden und nicht-weiterlernenden „AI systems“. Es gilt nur, die spezifischen Risiken für die weiterlernenden Systeme zusätzlich zu betrachten. Die Klassifizierung in „High-risk systems“ ist davon unabhängig.
D.h. der AI Act gilt für beide Typen.
Beste Grüße, Christian Johner
Liebes Johner-Team,
vielen Dank für den hilfreichen Artikel zur KI-Verordnung. Wir sind uns unsicher, was den Geltungsbeginn für Nicht-Medizinprodukte mit KI (KI-Modelle mit allgemeinem Verwendungszweck) betrifft. Wir konnten im Act die Stelle nicht finden, wo steht, dass die Anforderung an diese Produkte erst zum 02.08.2027 wirksam werden, falls diese vor dem 01.08.2025 in den Verkehr gebracht wurden. Können Sie uns hier auf die Sprünge helfen?
Vielen herzlichen Dank und Grüße,
Beatrice Dachsel
Danke für Ihre Nachfrage, Frau Dachsel!
Ich bezog mich auf den Artikel 111 (3), in dem es heißt:
„Anbieter von KI-Modellen mit allgemeinem Verwendungszweck, die vor dem 2. August 2025 in Verkehr gebracht wurden, treffen die erforderlichen Maßnahmen für die Erfüllung der in dieser Verordnung festgelegten Pflichten bis zum 2. August 2027.“
Ist diese Antwort hilfreich, oder habe ich Ihre Frage missverstanden oder einen Denkfehler gemacht?
Beste Grüße, Christian Johner
Vielen herzlichen Dank für Ihre zügige Hilfe! Aktuell sind wir noch am Klären, in welche „Kategorie“ unser Produkt nach dem AI Act fallen und welche der schrittweise geltenden Anforderungen auf uns zutreffen würden. Ggf. nutzen wir hierfür einmal Ihre MicroConsulting-Möglichkeit.
Viele Grüße, B. Dachsel
Hallo Herr Prof. Johner,
vielen Dank für die sehr gute Zusammenstellung.
Ich habe eine Frage bzgl. Artikel 111 (2) des AI Acts. Es gibt Diskussionen, dass dieser Artikel ein Schlupfloch für bereits im Markt befindliche (bzw. vor dem 02.08.2026 befindliche) Hochrisikosysteme ist. Der Artikel besagt ja, dass die gesamte Verordnung für bereits vor dem 02.08.2026 im Markt befindliche AI Systeme nur zur Anwendung kommt, „wenn an diesen Systemen ab diesem Zeitpunkt wesentliche Konstruktionsänderungen vorgenommen werden.“. Wenn man jetzt im Zuge der Verordnung davon ausgeht, das mit „Systemen“ das AI System gemeint ist, wäre man nicht betroffen, so lange man die AI im Medizinprodukt nicht wesentlich ändern würde?
Folgender Artikel hat mich zu dieser Annahme bewegt: https://www.hoganlovells.com/en/publications/the-ai-act-beyond-high-level-overviews-a-temporal-loophole
Ich freue mich auf Ihre Einschätzung.
Mit freundlichen Grüßen,
Anne Schmidt
Liebe Frau Schmidt,
danke für Ihre ausgezeichnete Frage!
Wie der von Ihnen verlinkte Artikel zeigt, scheinen sich auch die Juristen unsicher zu sein. Ich bin wegen des dort erwähnten „Loopholes“ etwas pessimistischer, weil der gekürzte Text lautet:
Ich lese daraus eine Ausnahme für die Betreiber, nicht für die Hersteller/Anbieter.
Ich habe noch ergänzende Gedanken:
Ich werde mich mit Juristen beraten und neue Erkenntnisse hier „posten“.
Nochmals besten Dank!
Herzliche Grüße, Christian Johner
Vielen Dank Herr Prof. Johner,
mir ist auch aufgefallen, dass im Deutschen Text von Betreiber gesprochen wird, im englischen und französischen Text an dieser Stelle aber der „operator/opérateur“ (Artikel 3 (8) der Definitionen) genannt ist.
Was die Sachlage nicht einfacher macht.
Ich freue mich auf Ihr Update.
Mit freundlichen Grüßen,
Anne Schmidt
In der Original-Sprachfassung des AI Acts wird der „operator“ genannt, bei dem es sich übersetzt um den in Art. 3 Nr. 8 definierten „Akteur“ handelt. In der deutschen Fassung wird in der Tat nur der „Betreiber“ im Sinne des Art. 3 Nr. 4 als Vorschriftsadressat genannt. Diese Abweichung in den verschiedenen Sprachfassungen führt zu einer erheblichen Diskrepanz des Adressatenkreises. Der Akteur umfasst einen weiten Personenkreis, zu dem unter anderem auch der Betreiber gehört. Der Betreiberbegriff ist demgegenüber ein wesentlich engerer. Der bindende deutsche Gesetzeswortlaut ist damit gegenüber den übrigen Sprachfassungen signifikant enger. Angesichts der unmittelbaren Geltung des für alle Mitgliedstaaten geltenden AI Acts ist davon auszugehen, dass es sich bei der Beschränkung auf den Betreiber in Art. 111 Abs. 2 um ein redaktionelles Versehen handelt. Dementsprechend muss sich der Geltungsbereich der Pflichten nach Art. 111 Abs. 2, abweichend vom Wortlaut, nicht nur auf Betreiber von Hochrisiko-KI-Systemen, sondern auf sämtliche Akteure in diesem Zusammenhang erstrecken.
So wohl die h.M. (vgl. Herold/Kehrberger, in: Bomhard/Pieper/Wende, KI-VO, Art. 111 Rn. 14).
Herzlichen Dank für Ihre wichtigen Hinweise, lieber Herr Dr. Hofer!
Danke auch für die Referenz!
Falls Sie Anregungen haben, wie ich den Artikel optimieren kann, um das stärker herauszustellen, freue ich mich darüber.
Beste Grüße, Christian Johner
Guten Tag an das Team des Johner Instituts
Vielen Dank für den Artikel.
Der Artikel erwähnt die Pflicht des Anbieters zur Registrierung in einer Datenbank (Art. 16 und 49).
Meine Analyse des relevanten Artikels (Art. 49(1)): Es werden lediglich in Anhang III aufgeführte Hochrisiko-KI-Systeme genannt, d.h. ein Medizinprodukt mit KI muss nicht nach AIA registriert werden, weil es nicht unter Anhang III fällt (sondern Anhang I nach Art. 6(1)).
Was meinen Sie?
Vielen Dank
L. Linnea
Sie haben völlig Recht, liebe Frau Linnea!
Ich habe in dem Kapitel 2.2. nicht klar differenziert gehabt zwischen Herstellern von KI-Systemen und Medizinprodukten. Daher habe ich jetzt klargestellt, dass – wie Sie richtig schreiben – die Medizinprodukte und IVD (und alle anderen Anhang-I-Produkte) nicht betroffen sind.
Herzlichen Dank für Ihren wertvollen Hinweis!
Beste Grüße, Christian Johner
Lieber Herr Johner
Vielen Dank für die rasche und klare Rückmeldung!
Sind Sie auch einverstanden mit meiner Analyse, dass die Anforderungen betr. ‚testing in real-world conditions‘ (Art. 60) nicht für KI-Systeme gelten, die MD/IVD sind oder als Sicherheitsbauteil in ein MD/IVD integriert sind? Sprich, dass pre-market Tests/Studien der genannten Produkte ’nur‘ den Anforderungen der MDR/IVDR (klinische Prüfungen mit MD / Leistungsstudien mit IVD) unterliegen.
Vielen Dank und beste Grüsse
L. Linnea
Sehr geehrte Frau Linnea,
die kurze Antwort ist: ja 🙂
Die längere Antwort ist: die Anforderungen des Artikels 60 betreffend weitgehend die KI-Systeme gemäß Anhang III, also keine Medizinprodukte. Er sagt auch, dass die „gegebenenfalls vorgeschriebene Ethikprüfungen unberührt“ blieben.
Letztlich will der Artikel 60 den Rahmen schaffen, dass man überhaupt „in der freien Wildbahn“ testen darf. Dieser Rahmen ist aber bei Medizinprodukten und IVD bereits gegeben. Daher ist dieser Artikel für die MP- und IVD-Hersteller nicht relevant.
Beste Grüße, Christian Johner
Guten Tag Herr Johner
Das macht Sinn. Herzlichen Dank für die Antwort.
Ich sehe den Punkt, dass der Rahmen bei Medizinprodukten und IVD bereits gegeben ist und der Artikel daher nicht relevant ist. Dennoch sind Anforderungen wie eine maximale Testdauer von sechs Monaten oder die Forderung, dass Vorhersagen, Empfehlungen oder Entscheidungen effektiv rückgängig gemacht bzw. außer Acht gelassen werden können, Aspekte, die die MDR IVDR nicht kennt (und bei denen man sich fragen kann, inwiefern es Sinn macht/ob das so gewollt ist, dass diese nur für Annex III Produkte gelten).
Beste Grüsse
L. Linnea
Guten Morgen –
im Abschnitt 5 steht (nun?):
„Artikel 64 der KI-Verordnung verlangt von den Herstellern, den Behörden einen vollständigen Remote-Zugriff auf die Trainings-, Validierungs- und Testdaten zu verschaffen, sogar durch eine API.“
Art.64 ist was ganz anders – aber auch in 21, 23, 72 ff. kann ich keine solche Regelung finden. Was ist damit gemeint?
Viele Grüße – HMMüller
Sehr geehrter Herr Müller,
danke für Ihren wichtigen Hinweis. In der Tat hat sich zur letzten Ausgabe des AI Acts nochmals einiges geändert.
Jetzt findet sich die Anforderungen im Anhang IV zur Technischen Dokumentation. Dort heißt es im Abschnitt 4.:
Dank Ihres Hinweises kann ich die Stelle im Beitrag aktualisieren. Nochmals besten Dank!
Viele Grüße, Christian Johner
Herzlichen Dank für diese kompakte Übersicht, die bei der Einordnung des Thema sicherlich für viele Akteure hilfreich ist.
Im Abschnitt 1.1.4 wird kurz auf Anforderungen aus Artikel 50 an die „bestimmten KI-Systeme“ eingegangen und mit dem Hinweis abgeschlossen, die Anforderungen seien durch die MDR bzw. IVDR im Wesentlichen bereits abgedeckt. Spezifische GSPRs zur Interaktion mit KI-Systemen oder der Kennzeichnung KI-generierter Inhalte bieten MDR/IVDR aber nicht, daher nehme ich an, Sie denken hier an allgemeine MDR/IVDR-Anforderungen zum sicheren und bestimmungsgemäßen Gebrauch bzw. an die vom Hersteller gelieferten Informationen.
Ich denke allerdings, dass der Artikel 50 durchaus knifflig in der Umsetzung sein kann. So benötigt man z. B. beim Artikel 50 (2) erstmal ein Verständnis darüber, was man als KI-generierte Inhalte auffasst (und was nicht) und ob eine der Ausnahmen greifen („Diese Pflicht gilt nicht, soweit die KI-Systeme eine unterstützende Funktion für die Standardbearbeitung ausführen …“). Im Kontext eines Medizinproduktes ist hier erstmal nicht klar, wie diese Begrifflichkeiten zu verstehen sind. Wann hört z. B. KI-generierter Inhalt, wie etwa Pixeldaten, nach menschlicher Interaktion und Veränderung auf, KI-generiert Inhalt zu sein? Was ist denn beim Medizinprodukt mit seiner spezifischen Zweckbestimmung als „unterstützende Funktion für die Standardbearbeitung“ zu verstehen (und was nicht)? Die Kommission arbeitet ja an Leitlinien zu den Transparenzanforderungen, ich warte mit Spannung auf deren Veröffentlichung.
Hat man dann mal (für sich) die nötige Klarheit erlangt, stellen sich die Fragen der Implementierung. Bleiben wir mal beim Artikel 50 (2). Falls der Hersteller zum Schluss kommt, im Sinne der Transparenz etwas tun zu müssen um auf KI-generierte Inhalte hinzuweisen, könnte dies einen „design change“ bedeuten und somit eine Änderung am Produkt – die dann natürlich zu bewerten ist, im Sinne der Medizinprodukteverordnung.
Das alles übrigens bis zum 2. August 2026, denn ab dann gilt der Artikel 50 (2), unabhängig von der Einstufung eines KI-Systems als „Hochrisiko-KI-Systems“ oder nicht. Dessen muss man sich zumindest bewusst sein, denn viele arbeiten vermutlich auf den 2. August 2027 hin (mit Hinblick auf die CE-Markierung auch sicherlich der Stichtag, an dem noch mehr „dranhängt“).
Je nachdem wie Ihre Sichtweise zu diesem Thema ist, wäre eventuell eine Ergänzung im Abschnitt 1.1.4 für die Leser hilfreich.
Nochmals vielen Dank.
Viele Grüße,
Martin Meyer
Das sind großartige Überlegungen und Anregungen, lieber Herr Meyer!
Vielen Dank!
Ich werde das in den nächsten Wochen „durch-deklinieren“ und im Journal als Praxistipp kommunizieren bzw. diesen Artikel ergänzen.
Nochmals vielen Dank! Sie helfen mir, den Artikel zu verbessern.
Herzliche Grüße, Christian Johner