
Übersetzungen und Begriffsdefinitionen
Was verseht man unter „Design“?
Im angloamerikanischen Sprachraum spricht man — so auch in der ISO 9001 bzw. ISO 13485 von „Design und Development“, was häufig als „Design und Entwicklung übersetzt wird. Damit ist die Frage, was mit Design gemeint sein kann, noch immer nicht beantwortet.
In der Schweizer Norm wird „Design und Development“ aufgrund des Mehrheitsbeschlusses im D-A-CH Ausschuss mit „Entwicklung“ übersetzt.
Wenn man Design überhaupt noch von der Entwicklung trennen will, würde es bei Software am ehesten dem Entwurf im Sinne einer Software-Architektur entsprechen.
Design Input und Design Output
Die „Design and development outputs“ werden in der ISO 13485 folgerichtig mit Entwicklungsergebnisse übersetzt, die „Design and development inputs“ mit „Entwicklungseingaben“, was eher eine schreckliche Übersetzung darstellt.
Definition der FDA
Die FDA gibt uns in 21 CFR part 820.3 (g) eine Definition:
„Design output means the results of a design effort at each design phase and at the end of the total design effort. The finished design output is the basis for the device master record. The total finished design output consists of the device, its packaging and labeling, and the device master record.“
Regulatorische Anforderungen an den Design Output
Forderungen der FDA
Im 21 CFR part 820.30(d) wird gefordert:
Each manufacturer shall establish and maintain procedures for defining and documenting design output in terms that allow an adequate evaluation of conformance to design input requirements. Design output procedures shall contain or make reference to acceptance criteria and shall ensure that those design outputs that are essential for the proper functioning of the device are identified. Design output shall be documented, reviewed, and approved before release. The approval, including the date and signature of the individual(s) approving the output, shall be documented.
Forderungen der ISO 13485/ISO 9001
Die Normen fordern in Kapitel 7.3.3 vom Design Output, dass er
- so vorliegen muss, dass man mit ihm prüfen kann, ob der Design Input damit erfüllt ist
- die Charakteristiken festlegt, die für den sicheren Gebrauch des Produkts wesentlich sind,
- freigeben wird und
- so formuliert ist, dass die Produktion, Beschaffung und Wartung wissen, was sie tun müssen, um das Produkt zu vervielfältigen (produzieren) und zu prüfen.
Und was ist nun Inhalt des Design Outputs?
Überlegungen der FDA
Die FDA gibt in ihrem Guidance Document „Design Control Guidance For Medical Device Manufacturers“ zu, dass nicht alle während der Entwicklung entstehenden Artefakte zum Design Output zählen und sagt, dass generell die Artefakte dazu gehören, die im Entwicklungsplan genannt sind und nennt sogar konkrete Beispiele wie Blockdiagramme, Flussdiagramme, „Software High-Level Code“ (dazu gleich mehr), sowie System und Subsystem-Spezifikationen. Damit kann der Design Output einer Phase oder Aktivität gleichsam wieder Design Input der nächsten sein.
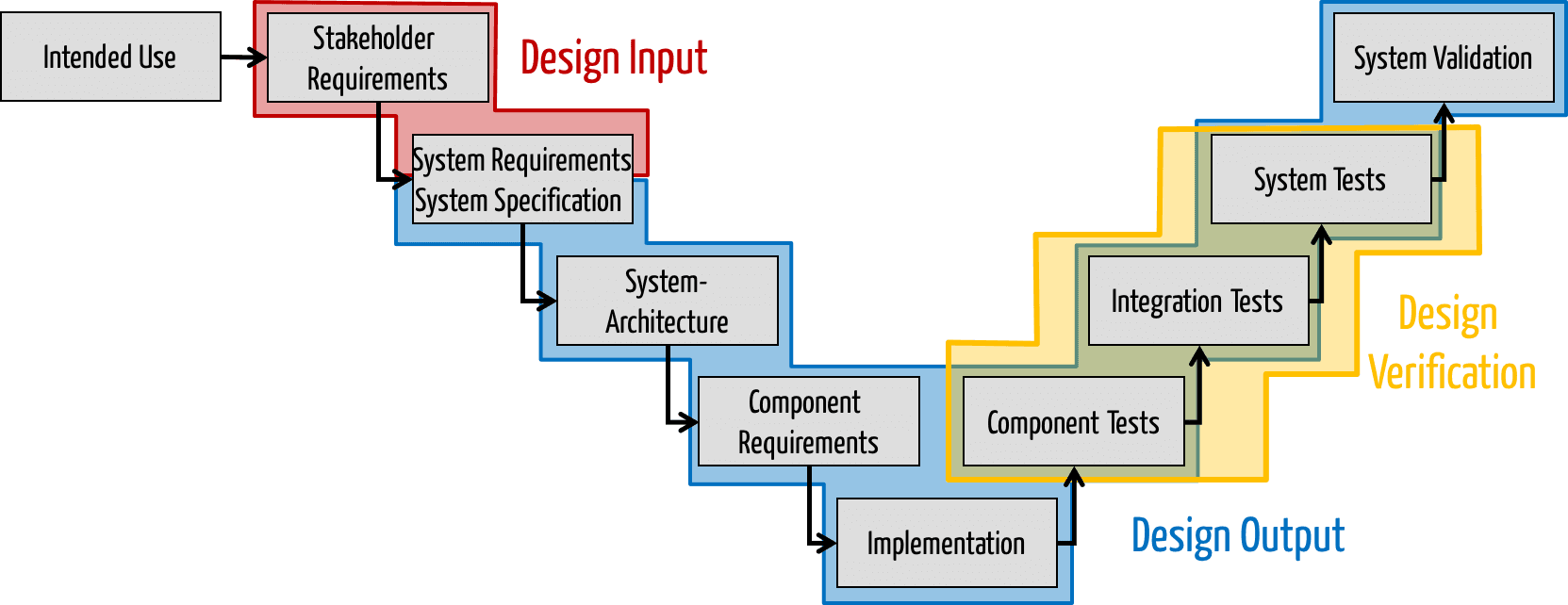
Die FDA zählt „Production Specifications“ dazu, also alles was festlegt, wie das Gerät zusammengebaut werden soll, Prozessspezifikationen, Installations- und Serviceanleitungen, Spezifikationen für Verpackung und Beschriftung.
Zu den „anderen beschreibenden Materialien“, die Bestandteil des Design-Outputs sein sollen, nennt die FDA explizit
- Ergebnisse der Risikoanalyse
- Software Source Code
- Ergebnisse der Verifizierungsaktivitäten.
Verwirrend mag manchmal sein, dass die FDA auch noch eine Design Verification und Design Validation kennt. Die FDA schreibt dazu „In design verification, the organization obtains objective evidence (i.e., data) that design outputs meet design inputs.“
Hier ist die FDA nicht ganz konsistent. Die Spezifikationen der Verifizierungsaktivitäten (z.B. Testspezifikation) lassen sich noch dem Design-Output zuordnen, die Verifizierungsergebnisse, also die Aufzeichnungen, sollten aber nur Teil der Verification sein. Andernfalls müsste man nochmals die Verifikation verifizieren. Letztlich ist diese Unschärfe auch nicht weiter tragisch. Zum einen kenne ich keinen Fall, wo ein falsche Aufteilung ein Problem im Audit (Inspektion) gewesen wäre, zum anderen landen sowieso alle Dokumente im Design History File.
Artefakte des Design Outputs
Für uns, die einen Schwerpunkt auf aktiven Medizinprodukten, die Software enthalten, haben, ergibt sich somit obiges Bild. Zu den Artefakten würden folglich zählen:
- Präzise Spezifikation der Benutzungsschnittstelle und Datenschnitttstellen (eine Diskussion, wann diese zum Design Input zählen finden Sie hier)
- System- und Software-Architektur
- Nur bei embedded Systemen: Software-Anforderungen
- Konstruktionszeichungen, Baupläne,
- Stückteilelisten
- Bestückungspläne
- Verpackung und sonstiges Labeling
- Verifizierungs- und Validierungspläne und zugehörige Ergebnisse
- Software-Code
- Code-Review-Protokolle, Checklisten und andere Dokumentenfreigaben
- Ergebnisse der statischen Code-Analyse
Achten Sie darauf, dass Sie diese Dokumente und Aufzeichnungen lenken und deren Versionen für verschiedene Entwicklungsstände konsistent zuordnen können. Andernfalls verletzen Sie die Anforderungen an das Design History File.


