
Die FDA hat ein Interoperability Guidance als Entwurf veröffentlicht. Es trägt den Titel „Design Considerations and Pre-market Submission Recommendations for Interoperable Medical Devices“ (hier zum Download).
Lesen Sie hier, was die FDA damit bezweckt und auf welche Anforderungen Sie sich einstellen müssen.
Interoperability Guidance Document: Was die FDA bezweckt
#1 Eigene Sicht darstellen und damit Forderungen formulieren
Die FDA möchte mit diesem Dokument Klarheit darüber verschaffen, wie sie Medizinprodukte bei Einreichungen bewertet, die mit anderen Systemen elektronisch kommunizieren und interagieren. Letztlich formuliert die FDA damit Ihre Anforderungen an die Interoperabilität.
#2 Risiken durch vernetzte Medizinprodukte minimieren
Die US-Gesundheitsbehörde hat erkannt, dass Medizinprodukte immer stärker vernetzt werden – mit Medizinprodukten und Nicht-Medizinprodukten wie klinischen Informationssystemen. Ihr ist auch bewusst, welche Chancen, aber auch Risiken damit verbunden sind. Letztere möchte sie minimieren.
#3 Interoperabilitäts-Standards fördern
Die FDA glaubt, dass (nur) durch Interoperabilitätsstandards sichere und zuverlässige interoperable Systeme geschaffen werden können. Daher setzt sie sich dafür ein – man könnte auch sagen, daher möchte sie diese Standards erzwingen.
Was die FDA von Ihnen erwartet
Die FDA wünscht sich in ihrem Interoperability Guidance Document, dass Sie bei der Entwicklung von Systemen, die über eine elektronische Datenschnittstelle (z.B. TCP/IP Socket, USB, wireless oder kabelgebunden) laufen, einige Überlegungen anstellen.
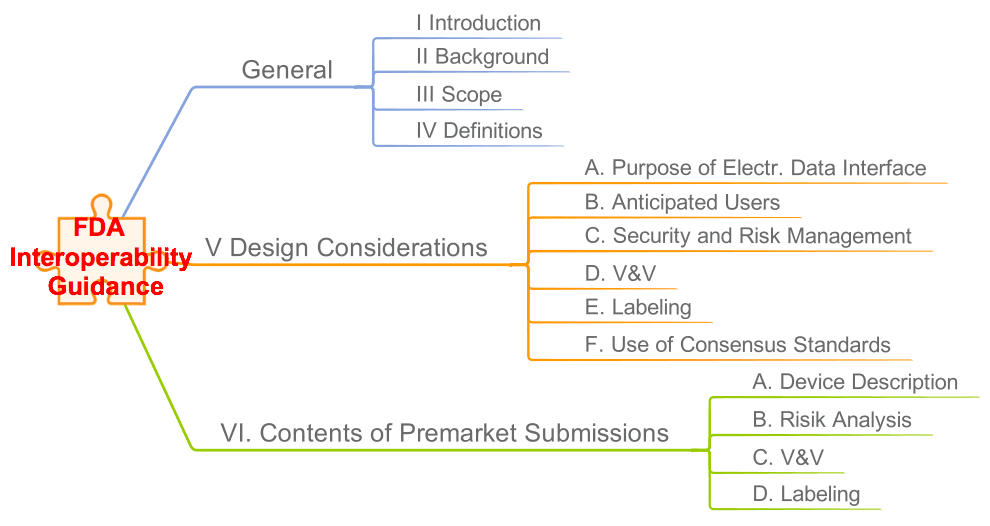
#1 Zweckbestimmung festlegen
Sie sollen beschreiben,
- mit welchen Systemen Ihr Medizinprodukt Daten austauschen soll,
- was für ein Datenaustausch das ist (Senden, Empfangen, Steuerung, Alarme, Kommandos),
- welche Standards, Protokolle und Formate Sie nutzen (also alle Interoperabilitätsebenen adressieren!),
- welche funktionalen Performance-Anforderungen Sie stellen,
- was die Anwender mit diesen Daten tun können sollen und welche Kontraindikationen bestehen
- in welchem klinischen Kontext dieser Austausch passieren soll (z.B. ob die Infusionspumpe bei einem narkotisierten Patienten angeschlossen ist),
- welche Interoparabilitätsszenarien damit unterstützt werden sollen (also der ganze Datenfluss und dessen Zweckbestimmung).
Unser Tipp: Legen Sie kein weiteres Dokument an. Das sind Informationen, um die Sie Ihre Zweckbestimmung oder Ihre System-Spezifikation ergänzen sollten.
#2 Vorgesehene Anwender spezifizieren
Legen Sie fest, welche Anwender Sie vorsehen. Das sind nicht nur die primären Anwender, sondern auch die sekundären wie System-Integratoren oder Medizintechniker. Diese Personengruppen und deren Fähigkeiten sollten Sie auch im Rahmen des Risikomanagements betrachten.
Unser Tipp: Auch diese Festlegungen packen Sie am besten in bestehende Dokumente, insbesondere die Zweckbestimmung.
#3 Sicherheits- und Risikomanagement-Überlegungen anstellen
Erwartungsgemäß verlangt die FDA im Interoperability Guidance Document, dass Hersteller die Datenschnittstellen im Risikomanagement berücksichtigen. Dabei sollen Sie u.a. Folgendes betrachten:
- Hat die Datenschnittstelle (deren Existenz oder deren Implementierung) einen nachteiligen Einfluss auf die Sicherheit oder bestehende risikominimierende Maßnahmen?
- Müssen zusätzliche Sicherheitsmaßnahmen implementiert werden?
- Kann das Gerät mit fehlerhaften Daten umgehen?
- Welche Fehler können im Datenaustausch auftreten? Z.B. durch
- den Anschluss von geplanten und ungeplanten Systemen
- ungültige Kommandos oder Daten (eingehend und ausgehend)
- das Nicht-Erfüllen von Anforderungen z.B. an die Performanz?
- Können bestehende Standards dazu beitragen?
Unser Tipp: Dokumentieren Sie diese Überlegungen in Ihrer Risikomanagementakte.
#4 Verifizierung und Validierung
Auch die folgenden Anforderungen der FDA überraschen nicht: Stellen Sie sicher, dass die Maßnahmen, die Sie festgelegt haben, um Risiken zu minimieren, auch wirksam sind. Zu diesen Maßnahmen zählen:
- Fähigkeit mit Daten umzugehen, die korrupt oder außerhalb der Spezifikation sind.
- Fähigkeit, grundlegende Anforderungen und wesentliche Leistungsmerkmale immer zu erfüllen.
- Erfüllung von Consensus-Standards (dazu gleich mehr).
- Sicherstellung, dass nur autorisierte Anwender und Systeme sich mit dem Medizinprodukt verbinden können.
- Gebrauchstaugliche Benutzerschnittstelle – um Datenschnittstellen zu parametrieren? Das wird nicht ganz klar.
#5 Labeling
Informationen zu Risiken und zum richtigen Gebrauch der Schnittstellen müssen z.B. in der Gebrauchsanweisungdokumentiert sein.
#6 Consensus Standards
Die FDA sagt im Interoperability Guidance Document, dass sie einige „Consensus Standards“ bereits anerkannt habe, weil sie deren Nutzen erkennt. Sie nennt im Dokument keine konkreten nennt aber die Themen:
- Datenformate
- Interoperabilitäts-Architektur (was auch immer das ist)
- Andere Aspekte der Interoperabilität
Allerdings verlinkt die FDA auf ein weiteres Dokument, das diese Standards nennt. Dazu zählen:
- IEC 80001-Familie
- ISO/IEC 11073-Familie (für Point of Care Geräte)
- IEC 62344 Network and System Security
Unser Tipp: Legen Sie die Interoperabilitäts-Standards bereits in der System- bzw. Software-Requirements-Specification fest.



Hallo,
könnte man sich diesen Anforderungen nicht einfach durch den Kunstgriff entziehen, indem man die Konnektivität (z.B. BT) erst gar nicht in den bestimmungsgemäßen Gebrauch mit aufnimmt, stattdessen die Spezifikationen der Daten kontrolliert (Vertrag) weitergibt und damit auch die Verantwortung für die medizinische Verwendung der Daten weitergibt ?
VG
JB
Der Beitrag war noch gar nicht fertig. Sorry, dass er schon live zu sehen ist/war.
Genau diesen Kunstgriff möchte die FDA vermeiden. Zum einen kann und darf man nicht Teile des bestimmungsgemäßen Gebrauchs unter den Tisch fallen lassen. Zum anderen enthält das System Komponenten, die die Schnittstellen implementieren. Diese müssen im Risikomanagement betrachtet werden. Damit ist man wieder beim vollen Umfang dieses Guidance Documents angekommen.