
Die GLP (Good Laboratory Practice) definiert Anforderungen an ein Qualitätssicherungssystem für nicht-klinische gesundheits- und umweltrelevante Sicherheitsprüfungen. Durch die GLP werden der organisatorische Ablauf und die Bedingungen festgelegt, unter denen Laboruntersuchungen geplant, durchgeführt und überwacht werden. Die Aufzeichnung und Berichterstattung der Prüfungen ist ebenfalls Gegenstand der GLP.
Lesen Sie in diesem Artikel, welche Anforderungen ggf. auch Medizinproduktehersteller erfüllen müssen, welche Regularien dabei zu beachten sind und welche dieser Anforderungen die Software betreffen.
GLP – Good Laboratory Practice: Was ist das?
Ziele der guten Laborpraxis
Die GLP soll die Qualität der Ergebnisse nicht-klinischer Laborstudien sicherstellen. Das betrifft insbesondere Laborstudien, die die Sicherheit folgender Produktekategorien nachweisen sollen:
- Arzneimittel
- Medizinprodukte
- Lebensmittelzusatzstoffe
- Pflanzenschutzmittel
- Kosmetische Artikel
- Elektronische Produkte
In verschiedenen Rechtsbereichen (z.B. EU, USA/FDA) unterscheidet sich diese Liste geringfügig.
Die Untersuchungen betreffen nicht (nur) die genannten Produkte selbst, sondern vor allem die „Prüfgegenstände“, die in diesen Produkten enthalten sind.
Beispiele für Laboruntersuchungen
Beispiele für solche Untersuchungen, die wohlgemerkt nicht an Menschen stattfinden, sind:
- Prüfung zur Bestimmung physikalisch-chemischer Eigenschaften, Inhalte, Konzentrationen
- Prüfung zur Bestimmung toxikologischer Eigenschaften (in vitro und in vivo)
- Prüfung zur Bestimmung erbgutverändernder Eigenschaften (in vitro und in vivo)
- Ökotoxikologische Prüfung (Auswirkungen auf aquatische und terrestische Organismen und natürliche Ökosysteme, Verhalten im Boden, im Wasser und in der Luft)
- Prüfungen zur Bestimmung von Rückständen
- Analytische Prüfungen an biologischen Materialien
Abgrenzung zur GMP (Good Manufacturing Practice) und GCP (Good Clinical Practice)
Die Anforderungen der GLP betreffen nicht die klinischen Studien an Menschen oder Tieren. Diese klinischen Studien unterliegen u.a. den GCP-Anforderungen.

Die Good Laboratory Practice haben auch nichts mit der Herstellung des Produkts zu tun. Hierfür sind die GMP-Richtlinien anzuwenden.
Anforderungen der Good Laboratory Practice GLP
Die GLP beschreibt ein Qualitätssicherungssystem, das einerseits übliche Anforderungen an Qualitätsmanagementsysteme beinhaltet wie die Verantwortung der Leitung und die Dokumentenlenkung, andererseits die Spezifika der Laborpraxis umfasst.
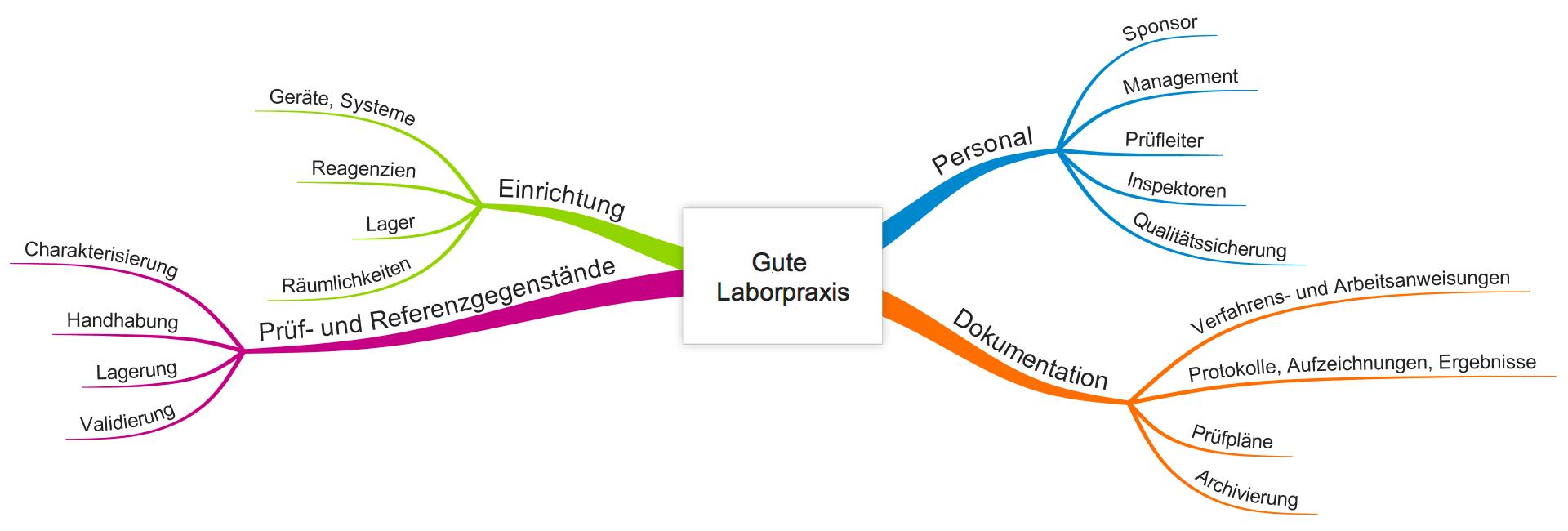
Regulatorische Anforderungen an die GLP
a) Wen die GLP (gute Laborpraxis) betrifft
Die regulatorischen Anforderungen sind nicht auf die pharmazeutische Industrie, medizinische Labore oder die Medizinproduktehersteller beschränkt. Alle Unternehmen, die o.g. Produktkategorien bzw. „Prüfgegenstände“ herstellen bzw. in den Markt bringen, unterliegen diesen regulatorischen Anforderungen.
Medizinproduktehersteller können davon in zweifacher Hinsicht betroffen sein:
- Eigene Produkte
Ihre eigenen Produkte müssen die grundlegenden Anforderungen der Medizinprodukterichtlinien erfüllen. Prüfungen von deren Eigenschaften und deren Unbedenklichkeit für die menschliche Gesundheit fallen ggf. unter die GLP. - Produkte für Labore und Prüfeinrichtungen
Insbesondere IVD-Hersteller liefern ihre Produkte an Prüfeinrichtungen. Die GLP stellt Anforderungen auch an die Geräte wie die Validierung computergestützter Systeme.
b) Übersicht über die Regularien
Die wesentlichen Anforderungen an die Good Laboratory Practice GLP formulieren die OECD Konsensus-Dokumente. Deren Anforderungen haben viele nationale und internationale Regulierungen übernommen.
USA
In den USA ist v.a. der 21 CFR part 58 („Good Laboratory Practice for Nonclinical Laboratory Studies“) zu nennen.
Die FDA hat auf ihrer Webseite einen Vergleich des 21 CFR part 58 und der OECD-Anforderungen veröffentlicht.
Europa
Auf europäische Ebene gibt es zwei Richtlinien, die Anforderungen an die GLP festlegen:
- Die Richtlinie 2004/9/EC verpflichtet die EU-Staaten, nationale Behörden zu bestimmen, die GLP-Inspektionen durchführen. Diese Inspektionen müssen den OECD-Richtlinien „Compliance Monitoring Procedures for GLP“ und „Conduct of Test Facility Inspections and Study Audits“ folgen. Diese Richtlinie löst die Richtlinie 88/320/EEC ab.
- Die Richtlinie 2004/10/EC verpflichtet die EU-Staaten, alle Maßnahmen zu ergreifen, dass die zertifizierten Laboratorien Prüfungen in Übereinstimmung mit den OECD-Richtlinien durchführen. Diese Richtlinie ersetzt die Richtlinie 87/17/EEC.
Europäische Nationalstaaten (z.B. Deutschland)
Die Nationalstaaten haben diese europäischen Richtlinien in nationale Gesetze überführt wie das Chemikaliengesetz und die Verwaltungsvorschrift GLP (ChemVwV-GLP) im Falle Deutschlands.
In Deutschland ist das Bundesinstitut für Risikobewertung, genau genommen „die GLP-Bundesstelle im BfR auf der Basis der „Allgemeinen Verwaltungsvorschrift GLP (ChemVwV-GLP)“ zuständig für die Koordinierung und Harmonisierung GLP-relevanter Fragen im nationalen und internationalen Bereich sowie in der Überwachung bestimmter GLP-Prüfeinrichtungen im In- und Ausland.“
Das Chemikaliengesetz benennt die gesetzlichen Anforderungen an die Good Laboratory Practice in Deutschland. Die Anforderungen beziehen sich daher auf Stoffe und Stoffgemische und nicht speziell auf Medizinprodukte.
Normen
Speziell für medizinische(!) Laboratorien bzw. Prülabore formulieren die ISO 15189 und die ISO/IEC 17025 Anforderungen. Häufig sind Labore nach beiden Normen akkreditiert.
Besondere Anforderungen der GLP an Software
Die regulatorischen Anforderungen an die Good Laboratory Practice GLP betreffen auch den Einsatz von Software und computergestützten Systeme.
a) USA / FDA
Sobald papierbezogene Dokumente und Aufzeichnungen durch computergestützte Systeme ersetzt werden, sind in den USA die Anforderungen des 21 CFR part 11 zu erfüllen.
Lesen Sie hier einen ausführlichen Artikel zum 21 CFR part 11.
Der Anwendungsbereich des FDA Software Validation Guidance Documents bezieht sich hingegen auf die Entwicklung und Herstellung von Medizinprodukten.
b) OECD zu computergestützten Systemen
Die OECD hat ein Konsensus-Dokument „Die Anwendung der Glp-Grundsätze auf Computergestützte Systeme“ veröffentlicht. Die Forderungen reichen von banal über nachvollziehbar bis absurd („Der Quellcode der Anwendungssoftware in der Prüfeinrichtung muss verfügbar oder durch diese abrufbar sein“).
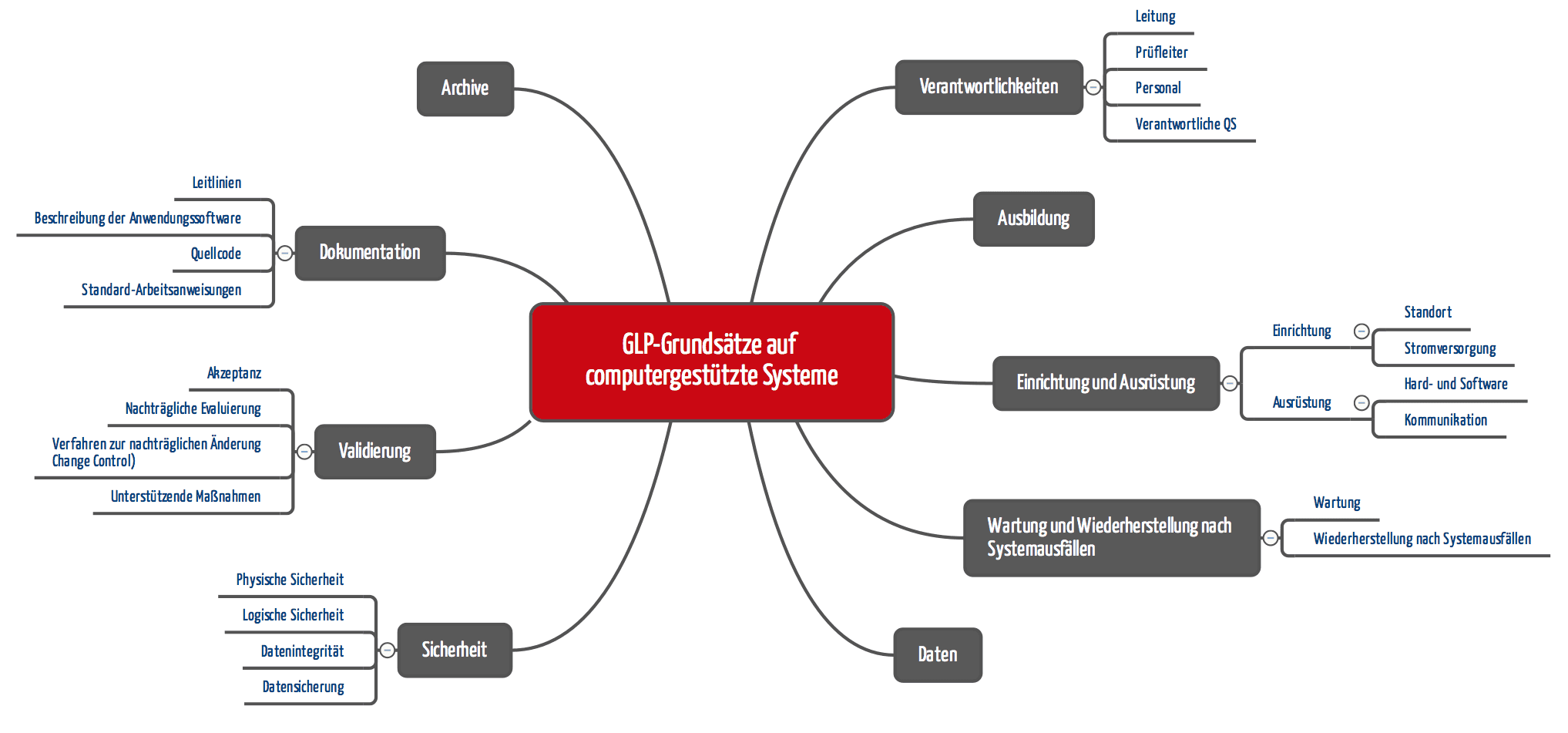
Auch für Medizinproduktehersteller sind die geforderten Verfahrensanweisungen interessant:
- Betrieb des computergestützten Systems (Hardware/Software)
- Sicherheitsmaßnahmen, um unbefugten Zugang und nicht genehmigte Programmänderungen zu bemerken und zu verhindern
- Änderung von Programmen und deren Dokumentation, Systemänderungen (Hardware/Software) einschließlich gegebenenfalls erforderlicher Tests vor der erneuten Inbetriebnahme
- Periodische Überprüfung der korrekten Funktion des gesamten Systems oder einzelner Komponenten und deren Dokumentation
- Wartungsverfahren computergestützter Systeme und der zugehörigen Ausstattung
- Softwareentwicklung und Anweisungen zur Durchführung von Akzeptanztests und deren Dokumentation
- Back-up für die gespeicherten Daten
- Desaster Recovery: Ausweichpläne zur Fortsetzung der Prüfung im Fall von Systemausfällen
c) EU-Richtlinien
In den EU-Richtlinien finden sich nur wenige Aussagen zum Umgang mit Software bzw. Computersystemen, von denen keine über die OECD-Vorgaben hinausgeht:
Die 2004/9/EC schreibt: The inspector should check […]
- that records have been kept of operation, maintenance, verification, calibration and validation of measuring equipment and apparatus (including computerised systems)
- in automated systems, data generated as graphs, recorder traces or computer print-outs are documented as raw data and archived.
- use, maintenance, cleaning, calibration and validation of measuring apparatus, computerised systems and environmental control equipment;
- any changes in the raw data, including data stored in computers, did not obscure previous entries, included the reason for the change and identified the person responsible for the change and the date it was made,
- computer-generated or stored data have been identified and that the procedures to protect them against unauthorised amendments or loss are adequate,
- the computerised systems used within the study are reliable, accurate and have been validated,
- records have been kept of operation, maintenance, verification, calibration and validation of measuring equipment and apparatus (including computerised systems),
Laut 2004/10/EC muss das Labor u.a. […]
- Verfahren einführen, die sicherstellen, dass computergestützte Systeme für ihre vorgesehene Anwendung geeignet sind und in Übereinstimmung mit diesen Grundsätzen der Guten Laborpraxis validiert, betrieben und gewartet werden.
- sicherstellen, dass im Verlauf einer Prüfung eingesetzte computergestützte Systeme validiert sind;
- Geräte, einschließlich validierter computergestützter Systeme, die zur Gewinnung, Erfassung und Wiedergabe von Daten und zur Kontrolle der für die Prüfung bedeutsamen Umweltbedingungen verwendet werden, zweckmäßig untergebracht werden, geeignet konstruiert und ausreichend leistungsfähig sind.
Die Forderungen gehen über die der 2004/9/EC nicht hinaus.
d) Nationale Vorschriften
Die nationalen Vorschriften übernehmen diese Forderungen.
In der Schweiz hat die Arbeitsgruppe Informationstechnologie am Bundesamt für Gesundheit einen Leitfaden für die Validierung computerisierter Systeme im GLP-Kontext entwickelt.
Lesen Sie hier mehr zum Thema Validierung computergestützter Systeme.
Fazit und Zusammenfassung
Die GLP (Good Labaratory Practice) definiert Anforderungen an QM-System von Laboreinrichtungen, die nicht-klinische Sicherheitsprüfungen von Produkten bzw. Substanzen durchführen. Diese Produkte sind nicht auf Medizinprodukte beschränkt.
Die OECD hat diese Anforderungen in mehreren Konsensus-Dokumenten formuliert. Die EU sowie die europäischen Nationalstaaten, ebenso die FDA orientieren sich stark daran und übernehmen teilweise die Anforderungen wörtlich.
Insbesondere IVD-Hersteller können die GLP-Richtlinien nutzen, um ihren Kunden auch Dienstleistungen anzubieten, um „GLP Compliance“ zu erreichen.
Versionshistorie:
- 2024-01-11: Überprüfung Aktualität + formale Änderungen/Ergänzungen
- 2016-12-20: Initiale Erstellung des Beitrags



Obwohl ich bereits auf langjährige Berufserfahrung als RA Manager in diversen Firmen und Funktionen zurückblicken darf, lerne ich immer wieder etwas dazu bei den verschiedenen Blog-Themen.
Immer wieder gehen weitere Türchen auf… und ich meine nicht diejenigen vom Weihnachtskalender.
Herzlichen Dank für die äusserst kompetenten und lehrreichen Blog-Themen und allen vom Johner Institut wünsche ich frohe Festtage und ein spannendes und erfolgreiches 2017!
Danke, ich freue mich sehr! Liebe Grüße Christian
1. Was wäre denn ein gutes Beispiel für Prüfungen von Eigenschaften des Medizinproduktes/dessen Komponenten die eindeutig unter GLP (insbesondere nach 21 CFR Part 58) fallen würden?
2. Gibt es GLP Anforderungen nach 21 CFR Part 58, die sich nicht aus GMP/21CFR 820/ISO 13485 ableiten lassen, d.h. darüber hinaus gehen?
(Messmittellenkung, Aufzeichnungslenkung, Prozessvalidierungen, u.ä. sind ja bereits auch ohne GLP gefordert)
Liebe Herr Thiemann,
vielen Dank für Ihre spannenden Fragen.
1. Ein Beispiel wären toxikologische Tests von Stoffen oder Stoffgemischen. Außerdem ist meine Empfehlung bei Tierversuchen darauf zu achten, dass diese auch nach GLP durchgeführt werden. z.B. in-vivo Biokompatibilitätstests. In der entsprechenden Norm ISO 10993-1 z.B. wird erwartet, dass die Qualität der bewerteten Daten sichergestellt ist. Dies kann über das Qualitätsmanagement System des beauftragten Labors erfolgen, welches entsprechend akkreditiert bzw. zertifiziert ist.
2. Der wichtigste Unterschied hier ist, dass die Anforderungen der GMP/21CFR820/ISO 13485 das Qualitätsmanagement der Hersteller betrifft und GLP/21 CFR Part 58 der Testlabore.