
Die Qualifizierung und die Klassifizierung von IVD-Software bestimmen, wie und wie schnell IVD-Hersteller ihre Software in den Markt bringen können und welche Kosten für die „Zulassung“ anfallen.
Dieser Artikel unterstützt Sie dabei, IVD-Software korrekt zu qualifizieren und zu klassifizieren. Damit können Sie regulatorische Probleme sowie daraus resultierende Aufwände und Verzögerungen vermeiden.
1. Begriffsklärungen
1.1 Qualifizierung
Unter Qualifizierung versteht man die Bewertung, ob ein Produkt unter die Definition des Begriffs Medizinprodukt bzw. In-vitro-Diagnostikum (IVD) fällt, oder eben nicht.
Das zweite Kapitel dieses Artikels beschreibt die Qualifizierung von IVD-Software.
1.2 Klassifizierung
Der Begriff der Klassifizierung ist doppelt belegt:
- Die Risikoklassifizierung von IVD bezeichnet die Einteilung in die Klassen A, B, C oder D gemäß Anhang VIII der EU-Verordnung 2017/746 über In-vitro-Diagnostika (IVDR).
- Daneben gibt es eine Software-Sicherheitsklassifizierung gemäß der Software-Norm IEC 62304, welche die Klassen A, B und C unterscheidet.
Das dritte Kapitel dieses Artikels beschreibt die Klassifizierung von IVD-Software, das vierte die Sicherheitsklassifizierung nach IEC 62304.
1.3 IVD-Software
Es gibt keine allgemeingültige Definition des Begriffs IVD-Software. Im Kontext dieses Artikels verstehen wir unter IVD-Software
- eine Standalone-Software-Anwendung,
- die als eigenständiges IVD qualifiziert ist (s. Kapitel 2),
- die als Zubehör zu einem IVD zählt, oder
- ein Software-System, das Teil eines IVD-Geräts ist.
Nicht zur IVD-Software zählen wir Standalone-Software-Anwendungen, die zwar im IVD-Kontext genutzt werden, aber
- in den Anwendungsbereich der EU-Medizinprodukte-Verordnung MDR fallen oder
- weder als Medizinprodukt noch als IVD zählen.
1.4 Medical Device Software – MDSW
Die MDCG-Leitlinie 2019-11 definiert den Begriff der Medical Device Software:
„Medical device software is software that is intended to be used, alone or in combination, for a purpose as specified in the definition of a “medical device” in the medical devices regulation or in vitro diagnostic medical devices regulation.“
Quelle: MDCG 2019-11
Damit sich eine Software als Medical Device Software (MDSW) qualifiziert, muss sie somit einen medizinischen Zweck aufweisen.
2. Qualifizierung von Software als IVD
Die MDCG 2019-11 Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 –MDR and Regulation (EU) 2017/746 – IVDR ist ein wertvoller Leitfaden zur Qualifizierung von medizinischer und nichtmedizinischer Software. Das Guidance-Dokument zerlegt die Qualifizierung in zwei Schritte:
- Bestimmung, ob die Software als MDSW zählt
- Bestimmung, unter welche EU-Verordnung (MDR oder IVDR) die Software bzw. das Produkt fällt
Die folgenden Teilkapitel stellen diese beiden Schritte vor.
2.1 Bestimmung, ob die Software als MDSW zählt
Im ersten Schritt müssen Hersteller prüfen, ob die Software unter die Definition des Begriffs „Medical Device Software” fällt. Das können sie ausschließen, wenn eine der folgenden Bedingungen zutrifft:
- Die Software macht nichts anderes, als Daten abzuspeichern, zu archivieren, zu kommunizieren oder eine einfache Suche darauf anzubieten.
- Die Software soll nicht individuellen Patienten einen (medizinischen) Nutzen bringen.
Falls die Software (als Standalone-Software oder als Teil eines Geräts) hingegen der Diagnose, Therapie, Überwachung oder Vorhersage von Krankheiten und Verletzungen dient, ist sie definitionsgemäß eine Medical Device Software (MDSW).
Damit zählen die Software oder das Gerät, dessen Teil die Software ist, als Medizinprodukt bzw. IVD und fallen unter den Anwendungsbereich der MDR bzw. IVDR.
Der Leitfaden MDCG 2019-11 beschreibt diesen ersten Schritt mithilfe einer Abbildung und Erläuterungen im Detail.
Lesen Sie mehr zur Einstufung, ob Software unter die EU-Medizinprodukte-Verordnungen fällt, in unserem Hauptartikel zu Software als Medizinprodukt.
2.2 Bestimmung, unter welche EU-Verordnung die Software fällt
Ob die Software bzw. das ganze Produkt die Anforderungen der MDR oder der IVDR erfüllen muss, hängt von mehreren Kriterien ab:
- Zweckbestimmung des Produkts
- Datenquellen für die angezeigten bzw. verarbeiteten Informationen
- Abhängigkeit der Zweckbestimmung von den Datenquellen
Der zweite Entscheidungsbaum in der Leitlinie MDCG 2019-11 erläutert die Fallunterscheidung (s. Abb. 1).
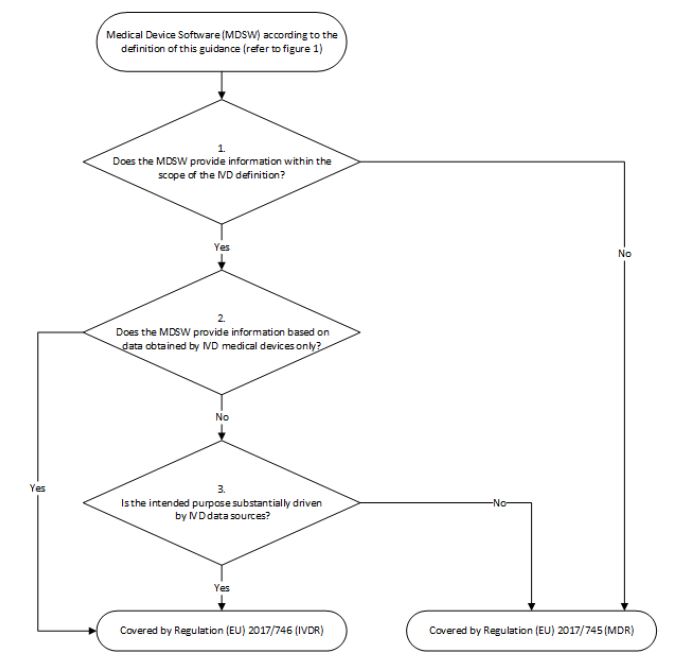
- Die erste Frage untersucht, ob die als MDSW qualifizierte Software anhand der IVD-Definition in den Anwendungsbereich der IVDR fällt. Wird diese Frage mit Nein beantwortet, ist die Software ausschließlich unter der MDR zu betrachten. Wird diese Frage mit Ja beantwortet, geht es weiter mit Frage 2.
- Fällt die Software unter die IVD-Definition, stellt sich die zweite Frage, ob die Input-Daten ausschließlich von einem IVD stammen. Wird diese Frage mit Ja beantwortet, fällt die Software unter die IVDR. Wird diese Frage mit Nein beantwortet, geht es weiter mit Frage 3.
- Die dritte Frage untersucht, ob die Software sowohl Daten aus „IVD-Quellen“ als auch aus anderen Quellen (z. B. von Medizinprodukten) nutzt. Es geht darum herauszufinden, ob die Zweckbestimmung der Software wesentlich durch IVD-Datenquellen erreicht wird. Wird diese Frage mit Ja beantwortet, fällt die Software unter die IVDR. Wird diese Frage mit Nein beantwortet, fällt die Software unter die MDR.
2.3 Weitere Interpretationen des MDCG-Dokuments
2.3.1 Kennzeichnung von Laborwerten, z. B. von Werten „out-of-range“
Ein System wie ein Laborinformationssystem wird nicht dadurch zu einer MDSW, dass es
- Laborwerte mit Labels versieht, beispielsweise um „out-ouf-range“-Werte zu kennzeichnen,
- Laborwerte filtert oder
- Daten an andere Systeme weiterleitet.
Komplexe Regeln und Berechnungen dienen in der Regel medizinischen Zwecken und führen zu einer Qualifizierung als Medizinprodukt oder IVD.
2.3.2 Parametrierung und Scripting
Wenn Hersteller den Anwendern die Parametrierung und ggf. sogar ein Scripting erlauben, die einen medizinischen Zweck haben, wird die Software (oder zumindest das jeweilige Modul) zur MDSW, falls das nicht bereits der Fall war. Die Organisation, die diese Anpassungen vornimmt, wird damit zum Eigenhersteller.
Allerdings führen einfache Berechnungen wie Einheitenumrechnungen noch nicht dazu, dass eine Software als Medizinprodukt oder IVD qualifiziert werden muss.
Lesen Sie hier, was Gesundheitseinrichtungen wie medizinische Labore bei der Entwicklung von Inhouse IVD, auch Laboratory Developed Test (LDT) genannt, beachten müssen
Beachten Sie weitere Hinweise im Borderline Manual der EU.

Meistern Sie Ihr IVDR-Zulassungsprojekt mit Leichtigkeit
Lernen Sie mithilfe unserer innovativen E-Learning-Plattform, wie Sie Ihr IVD sicher qualifizieren, wie die Klassifizierung von IVD unter der IVDR funktioniert,
wie Sie einen Plan zur Umsetzung der regulatorischen Anforderungen erstellen
und schlussendlich die Konformität Ihrer Akte prüfen können.
3. Risikoklassifizierung von IVD-Software gemäß IVDR
Die IVDR unterscheidet bei als IVD qualifizierten Produkten die Klassen A bis D. Dabei umfasst die Klasse A die unkritischen und die Klasse D die höchstkritischen Produkte.
3.1 Die Auswirkungen der Risikoklassifizierung
Die Klassifizierung wirkt sich nicht auf die Sicherheits- und Leistungsanforderungen aus, die die IVD-Software erfüllen muss. Selbst bei einer Software der Klasse A (nicht verwechseln mit der Klasse A gemäß IEC 62304) müssen alle anwendbaren Anforderungen gemäß Anhang I der IVDR erfüllt werden. Dazu gehören u. a.:
- Software-Lebenszyklus (IEC 62304)
- Risikomanagement (ISO 14971)
- Gebrauchstauglichkeit (IEC 62366-1)
- IT-Sicherheit
- Verifizierung und Validierung
Der Fachartikel zu den regulatorischen Anforderungen an IVD-Software nennt die grundlegenden Sicherheits- und Leistungsanforderungen an diese Produkte und die zugehörige Technische Dokumentation.
Die Klassifizierung entscheidet jedoch über die möglichen Konformitätsbewertungsverfahren: Bei IVD der Klasse A müssen die Hersteller keine Benannte Stelle involvieren, d. h. es findet keine Prüfung der Dokumente durch eine Benannte Stelle statt. Eine Behörde kann während der Marktüberwachung aber Einsicht in die Technische Dokumentation verlangen.
Zwar benötigen die Hersteller auch bei IVD der Klasse A ein QM-System, dieses muss aber nicht zertifiziert sein. Die Bewertung des QM-Systems für IVD der Klassen B bis D erfolgt automatisch während der Konformitätsbewertung gemäß Anhang IX der IVDR.
3.2 Klassifizierungsregeln gemäß IVDR
Die IVDR enthält vergleichbar der MDR u. a. die folgenden Klassifizierungsregeln:
1.1. Die Anwendung der Klassifizierungsregeln richtet sich nach der Zweckbestimmung der Produkte.
1.2. Wenn das betreffende Produkt dazu bestimmt ist, in Verbindung mit einem anderen Produkt angewandt zu werden, werden die Klassifizierungsregeln auf jedes Produkt gesondert angewendet.
1.3. Zubehör für ein In-vitro-Diagnostikum wird unabhängig von dem Produkt, mit dem es verwendet wird, gesondert klassifiziert.
1.4. Software, die ein Produkt steuert oder dessen Anwendung beeinflusst, wird derselben Klasse zugerechnet wie das Produkt. Ist die Software von anderen Produkten unabhängig, so wird sie für sich allein klassifiziert.
1.8. Wenn der Hersteller für ein Produkt mehrere Zweckbestimmungen angibt und das Produkt daher mehr als einer Klasse zuzuordnen ist, wird es in die höchste der Klassen eingestuft.
1.9. Für den Fall, dass für dasselbe Produkt mehrere Klassifizierungsregeln gelten, wird die Regel der Einstufung in die höchste dieser Klassen angewendet.
Quelle: IVDR, Anhang VIII
Die IVDR klassifiziert die Produkte unter Berücksichtigung der zugrundeliegenden Krankheitsbilder bzw. anhand der möglichen (kritischen) Auswirkung, die eine fehlerhafte Information, die durch ein IVD für einen Patienten / eine Person bereitgestellt wird, haben kann. Einer „Regel 11“ wie bei der MDR bedarf es daher nicht.
3.3 Anwendung der Klassifizierungsregeln
Da sich die Klassifizierung nach der Zweckbestimmung richtet, ist es entscheidend, ob die Standalone-Software medizinische Informationen über den Patienten bereitstellt, also die klinische Aussage erlaubt. Anders formuliert, ob die Spezifität und ggf. die Sensitivität des Analyten erst durch die Software hergestellt wird.
Bioinformatische Software, die genetische Daten aus einer Hochdurchsatz-Sequenzierung von DNA-Proben mit Referenzdaten vergleicht, um z. B.
- festzustellen, ob eine Krankheit vorliegt, oder
- einen bestimmten physiologischen Zustand zu diagnostizieren oder zu analysieren oder
- Stadien einer Krebserkrankung einzuteilen
fallen in Klasse C; es sei denn, es handelt sich um eine durch übertragbare Erreger verursachte “lebensbedrohende Krankheit mit einem hohen oder mutmaßlich hohen Verbreitungsrisiko”. Dann fällt die IVD-Software sogar in Klasse D.
3.3.1 IVD-Software in „Risikoklasse“ A?
Die Aussage, dass IVD-Software immer in Klasse A falle, weil Software den Patienten nie direkt schaden oder weil die Software selbst nichts tun könne, ist nicht korrekt.
Falls eine Software ein IVD-Instrument steuert, wird sie derselben Klasse wie das Instrument zugeordnet (gemäß Durchführungsvorschrift 1.4).
Wenn das Instrument Regel 5b) entspricht und keine analyt-spezifische Datenauswertung vornimmt, fällt es in Klasse A. Nur in diesem Fall fällt auch eine IVD-Software in Klasse A.
3.3.2 Risikoklassen B, C und D
Falls die IVD-Software assay-spezifische Auswertungen vornimmt, muss sie der Klasse des Assays zugeordnet werden. Die Software beeinflusst die Anwendung und liefert selbst die diagnostischen Informationen im Sinne einer medizinischen Zweckbestimmung.
Sollen verschiedene Assays mit der Software ausgewertet werden, sind auch die Durchführungsregeln 1.8 und 1.9 zu beachten (Klassifizierung in die höchste Klasse der betreffenden Assays!).
Es ist eine strategische Entscheidung, ob man Software in steuernde Elemente und auswertende Elemente trennt, um sie unterschiedlich klassifizieren zu können.
Nutzen Sie das Expertenteam des Johner Instituts bei der Festlegung Ihrer Segregationsstrategie bzw. Ihrer regulatorischen Strategie!
4. Sicherheitsklassifizierung von IVD-Software gemäß IEC 62304
4.1 Problemstellung
Ein Point-of-Care-Gerät misst „übliche“ Parameter wie pH, pCO2, Glukose, Kalium usw. Wie ist dessen Software gemäß IEC 62304 zu klassifizieren?
- Ein Team argumentiert für Sicherheitsklasse B, weil keine schwere Verletzung möglich sei. Schließlich sei das Messergebnis des IVD-Geräts nur einer von vielen Parametern für eine Therapie – eben die richtige oder die falsche oder eine richtige, aber verspätete. Zudem würden im Laborgerät regelmäßig Kalibrierungs- und Kontrollzyklen gefahren.
- Ein anderes Team argumentiert für Sicherheitsklasse C, weil es in einem unwahrscheinlichen Fall tatsächlich eine Patientenschädigung geben könnte. Die Ärzte als „risikominimierende Maßnahme“ ziehen sie dabei nicht in Betracht.
Welche Einschätzung stimmt?
4.2 Besonderheiten von IVD
Bei IVDs, genauer gesagt deren bereitgestellten Informationen, gibt es im Gegensatz zu anderen Medizinprodukten keine Möglichkeit einer direkten Schädigung: keine Quetschung, keine Strahlenschäden, kein elektrischer Schlag. Die Messwerte können „nur“ dazu führen, dass Patienten falsch, verzögert oder gar nicht behandelt werden – was je nach Kontext der Anwendung fatale Folgen haben kann.
Diese „äußere Ursachenkette“, also die Ursachenkette beginnend beim falsch, nicht oder verspätet angezeigten Messwert, ist schwierig abzuschätzen. Besonders schwer abzuschätzen sind die Wahrscheinlichkeiten – etwa dass ein Arzt tatsächlich aufgrund eines falschen oder fehlenden Messwerts falsch behandelt.
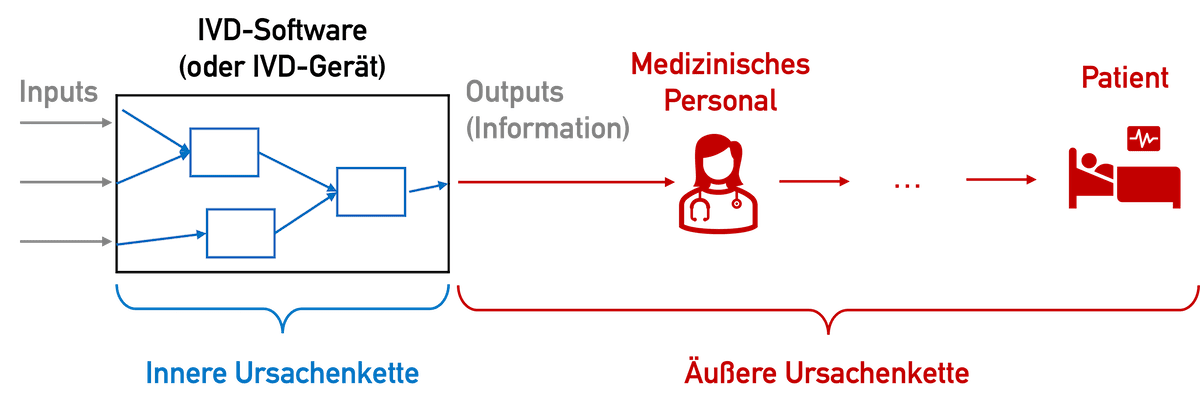
Leider betrachtet die IEC 62304 diese Wahrscheinlichkeiten nicht. Damit kann bei „ausreichend kritischen“ Patienten im ungünstigsten Fall ein schwerer Schaden eintreten. Damit ergibt sich die Sicherheitsklasse C. Die IEC 62304:2006 + A1:2015 gewährt hierbei mehr Optionen durch Maßnahmen außerhalb der Software.
Lesen Sie mehr zu den Software-Sicherheitsklassen nach IEC 62304.
Die Kalibrier- und Kontrollzyklen ändern meist nichts am Schweregrad; sie reduzieren nur die Wahrscheinlichkeit, dass ein Messwert falsch oder gar nicht angezeigt wird und zu einem Schaden führt. Wahrscheinlichkeiten sind aber für die Sicherheitsklassifizierung weitgehend irrelevant.
4.3 Die Zweckbestimmung entscheidet
Die Sicherheitsklasse hängt bei IVD, wie die Risikoklasse (s. Kapitel 3), v. a. von der Zweckbestimmung ab. Denn die Zweckbestimmung legt wiederum die Zielpopulation, also die Patienten bzw. deren Gesundheits- oder Krankheitszustand fest. Daraus ergeben sich der maximale Schweregrad des Schadens, der durch einen Softwarefehler verursacht werden kann und – weitgehend unabhängig von der Wahrscheinlichkeit – damit die Sicherheitsklasse der Software gemäß IEC 62304.
Die Zielpopulation wird auch durch die beabsichtigte Funktion des IVDs beeinflusst und muss betrachtet werden (z. B. ein Screening-Test in einer überwiegend gesunden Population oder ein Bestätigungstest auf Basis von Risikofaktoren oder Voruntersuchungen und somit an einer vorwiegend bereits erkrankten (Hochrisiko-)Population). Die Leistungsparameter (wie der positive und der negative prädiktive Wert) in der entsprechenden Bevölkerungsgruppe zeigen schließlich die Wahrscheinlichkeit, dass eine untersuchte Person auch das richtige Ergebnis erhält, auf.
Zur Bewertung der äußeren Ursachenkette bis zum Schaden am Patienten sollte darüber hinaus der Stand der Technik in der Medizin berücksichtigt werden, um zu eruieren, welche Therapieoptionen verfügbar sind und welche Patientenmanagement-Entscheidungen auf Basis der durch die IVD-Software bereitgestellten Informationen getroffen werden.
Die Antwort auf die in Kapitel 4.1. eingeleitete Frage ist demnach: Die Zweckbestimmung entscheidet.
Lesen Sie hier mehr zum Thema Software as Medical Device (SaMD).
5. Fazit und Zusammenfassung
Die Qualifizierung und Klassifizierung von IVD-Software bestimmen die Dauer und die Aufwände für die „Zulassung“ dieser Produkte.
Die Qualifizierung und Klassifizierung können die Hersteller jedoch beeinflussen durch die Zweckbestimmung sowie die Aufteilung des Produkts auf ein oder mehrere IVD, Zubehöre oder Nicht-IVD-Produkte.
Diese Festlegungen sind Teil der regulatorischen Strategie, die auch die Zulassung in anderen Ländern sowie die Post-Market-Phase beeinflussen. Entsprechend wichtig und wegweisend ist die regulatorische Strategie.
Das Johner Institut unterstützt IVD-Hersteller dabei, die regulatorische Strategie klug festzulegen, um unnötige regulatorische Aufwände zu vermeiden und die Inverkehrbringung der Produkte in möglichst kurzer Zeit zu ermöglichen.
Änderungshistorie
- 2024-03-26: Artikel komplett neu geschrieben
- 2019-10-30: Erste Version des Artikels veröffentlicht (wegen MDCG 2019-11)


Wie werden Apps behandelt?
Beispiel: Das IVD Ergebnis ist eine Färbung, die normalerweise visuell ausgewertet wird.
Nun soll eine App (auf dem Handy) die Auswertung machen.
Ist die App dann Zubehör oder ein eigenständiges IVD?
Sehr geehrte Frau Dr. Theiling,
das ist eine spannende Frage. Danke, dafür!
In diesem Fall haben Sie beide Möglichkeiten. Allerdings gibt es benannte Stellen, die Software nicht als Zubehör zulassen wollen. Wenn Ihr IVD bereits in der Zweckbestimmung enthält, dass die Färbung visuell oder per Software ausgewertet werden soll, wäre m.E. das Zubehör dennoch die richtige Klassifizierung. Ebenso wenn die App direkt zu/mit einem spezifischen IVD verwendet werden soll.
Regulatorisch ist der Unterschied minimal. In beiden Fällen bringen Sie wahrscheinlich ein Klasse I Produkt in Verkehr, wahrscheinlich auch ohne Messfunktion im Sinn der MEDDEV.
Beste Grüße, Christian Johner.
ReCaptcha test
Liebes Team,
Betreff: Zubehör von IVD Software
Frage bezieht sich auf eine eigenständige IVD Software die selbst ein IVD gemäß IVDR Klasse C wird, im moment jedoch noch nach IVDD zugelassen ist.
Zur Analyse wird eine menschliche Probe in ein Röhrchen mit Stabilisator vom Patienten selbst abgegeben, welches dann im Labor/Krankenhaus analysiert wird und die Daten in der IVD Software ausgewertet werden. Welchen Kriterien muss mein Probeentnahme Röhrchen entsprechen? Muss es auch Klasse C gemäß IVDR sein?
Freue mich über Inputs.
LG,Anna
Liebe Anna,
aus dem Betreff entnehme ich, dass das Probennahme-Röhrchen als Zubehör zur IVD-Software definiert ist. Das ist so leider nicht möglich, da Zubehör definiert ist als
Gegenstand, der zwar an sich kein In-vitro-Diagnostikum ist, aber vom Hersteller dazu bestimmt ist, zusammen mit einem oder mehreren bestimmten In-vitro-Diagnostika verwendet zu werden […]
Die IVD-Definition sagt jedoch ausdrücklich: Probenbehältnisse gelten als In-vitro-Diagnostika. Daher können Probenbehältnisse kein Zubehör sein.
ungeachtet dessen: Wenn Sie das Probennahmeröhrchen als eigenständiges Produkt mit eigener Zweckbestimmung definieren, können Sie die beiden Produkte für die Verbindung miteinander vorsehen und dennoch getrennt klassifizieren (siehe IVDR Anhang VIII 1.2.). Wenn das Probennahmeröhrchen zudem keine kritischen Charakteristika besitzt, die andere Regeln als Regel 5 anwendbar machen, dann würde das Probennahmeröhrchen in Klasse A landen (Regel 5 c).
Ich hoffe, ich konnte Ihnen damit weiterhelfen.
Mit meinen allerbesten Grüßen,
Sebastian Grömminger
Guten Morgen Johner-Team,
es handelt sich um ein Diagnostik-Gerät für die Eigenanwendung und eine Smartphone-App.
Ich benötige bitte eure Unterstützung bei folgendem Sachverhalt:
Was darf ein Patient, der das Diagnostik-Gerät zu Hause nutzt, dessen Daten auf eine Smartphone-App übertragen werden, auf dem Screen des Smartphones sehen – Werte und Empfehlungen oder nur Werte?
Hat die Handlungsempfehlung bzw. Interpretation eines Werts (z. B. Zielkorridor) in der App einen Einfluss darauf, ob es sich um zwei unabhängige Produkte (IvD = IVDR und App = MDR) handelt oder nicht?
Wenn ich richtig verstanden habe, ist es entscheidend, ob die Zweckbestimmung die Auswertung per Software (App) vorsieht. Richtig? Wären dann die App in diesem Fall ein Zubehör zu dieser IvD und beide Produkte über die IVDR geregelt?
Ich hoffe, ich konnte den Sachverhalt und die Fragestellungen verständlich formulieren.
Lieben Dank im Voraus und viele Grüße,
Mary
Liebe Mary,
der geschilderte Fall erscheint mir etwas zu komplex, um ihn über eine Antwort hier im Blog vollends beantworten zu können. Zwei Aussagen möchte ich dennoch machen:
1. Welche Aussagen angezeigt werden dürfen, hängt von der Zweckbestimmung, der Leistungsbewertung und der Gebrauchstauglichkeit ab, die für das Produkt belegt werden kann.
2. Wenn die App im wesentlichen IVD Datenquellen nutzt zur Interpretation der Ergebnisse und Ableitung von Handlungsempfehlungen, ist die App ein eigenständiges IVD unter IVDR oder besser gesagt eine IVD-Medical Device Software (IVD-MDSW). Mehr dazu finden Sie in der MDCG 2019-11.
Wenn wir Sie bei der Qualifizierung Ihrer Software-App unterstützen sollen, melden Sie sich gerne bei uns.
Mit herzlichen Grüßen,
Sebastian Grömminger
Liebes Johner-Team,
Vielen Dank für diesen ausführlichen Artikel. Ich hätte eine tiefergehende Frage: Nach Decision Step 2 der MDGC 2019-11 gilt eine Software ja stets als IVD-Software, wenn sie ausschließlich Daten von IVD-Medizinprodukten verarbeitet (bzw. wenn die IVD-Daten ausschlaggebender für die Zweckbestimmung als die MD-Daten sind).
In den Beispielen der MDGC ist dabei oftmals von Rohdaten die Rede. Macht es nun für die Einordnung als IVD/MD irgendeinen Unterschied, ob die von der Software verarbeiteten Daten bereits vorher bearbeitet, interpretiert wurden, also bereits Sekundärdaten darstellen?
z.B. ein Labor wertet mit einem Gerät eine Blutprobe aus und übergibt einen Befund. Diese Verarbeitung macht das Gerät wohl zweifellos zum IVD.
Ist nun eine App, die diesen Befund (d.h. bereits verarbeitete Daten) übernimmt und daraus wiederum neue Erkenntnisse zieht (angenommen, es handelt sich um eine medizinische Zweckbestimmung) wiederum als IVD zu qualifizieren?
Mit herzlichen Grüßen,
Helmut
Sehr geehrter Helmut,
danke für die wichtige Frage!
Die Verarbeitungskette von „Rohdaten“ bis zu den final ausgewerteten Daten umfasst meist viele Schritten. Zuerst im „Gerät“ selbst, später dann ggf. auf Plattformen oder/und schließlich in „End-Anwendungen“ wie einer App. Entscheidend ist nicht, wo in dieser Verarbeitungskette das Produkt einsortiert ist, sondern
In Ihrem Fall erscheint es somit so, dass Ihre App als IVD zu qualifizieren wäre.
Viele Grüße
Christian Johner