
Hersteller, die Machine Learning (ML) in ihren Medizinprodukten oder IVD verwenden, müssen viele regulatorische Anforderungen erfüllen.
Dieser Artikel gibt eine Übersicht über die wichtigsten Regularien und Best Practices für die Umsetzung. Er erspart Ihnen somit das Recherchieren und Lesen von hunderten Seiten und hilft Ihnen, sich perfekt auf das nächste Audit vorzubereiten.
- Die KI-/ML-spezifischen regulatorischen und normativen Anforderungen sind noch im Entstehen; sie sind inkonsistent und unvollständig und ändern sich rasch.
- Diese Anforderungen unterscheiden sich in den verschiedenen Märkten stark.
- Hersteller sollten zuerst die KI-/ML-unspezifischen Anforderungen (z. B. an die Validierung) auf Medizinprodukte mit Machine Learning übertragen.
- Der Leitfaden des Johner Instituts, auf dem der Questionnaire des TeamNB basiert, beschreibt in Europa den Stand der Technik und konsolidiert alle regulatorischen und normativen Anforderungen.
- Die Umsetzung der Anforderungen bedingt eine gute Kommunikation zwischen Regulatory Affairs und Quality Management einerseits sowie Data Scientists und Machine Learning Experts andererseits.
Dieser Schlagwort-Artikel zur künstlichen Intelligenz verschafft eine Übersicht und verlinkt weitere Fachartikel zum Thema.
1. Gesetzliche Anforderungen an den Einsatz von Machine Learning bei Medizinprodukten in der EU
a) EU-Medizinprodukterecht
Offensichtlich müssen auch Medizinprodukte mit Machine Learning die bereits bestehenden regulatorischen Anforderungen wie die MDR und IVDR erfüllen, z. B.:
- Hersteller müssen den Nutzen und die Leistungsfähigkeit der Medizinprodukte nachweisen. Bei Produkten, die der Diagnose dienen, bedarf es z. B. des Nachweises der diagnostischen Sensitivität und Spezifität.
- Die MDR verpflichtet die Hersteller, die Sicherheit der Produkte zu gewährleisten. Dazu zählt, dass die Software so entwickelt wurde, dass Wiederholbarkeit, Zuverlässigkeit und Leistungsfähigkeit gewährleistet sind (s. MDR Anhang I, 17.1 bzw. IVDR Anhang I, 16.1).
- Hersteller müssen eine präzise Zweckbestimmung formulieren (MDR/IVDR Anhang II). Sie müssen ihre Produkte gegen die Zweckbestimmung und die Stakeholder-Anforderungen validieren und gegen die Spezifikationen verifizieren (u. a. MDR Anhang I, 17.2 bzw. IVDR Anhang I, 16.2). Hersteller sind auch verpflichtet zur Beschreibung der Methoden, mit denen sie diese Nachweise führen.
- Basiert die klinische Bewertung auf einem Vergleichsprodukt, muss technische Äquivalenz gegeben sein, was die Bewertung der Software-Algorithmen explizit einschließt (MDR, Anhang XIV, Teil A, Absatz 3). Dies ist bei der Leistungsbewertung von In-vitro-Diagnostika (IVD) noch weitaus schwieriger. Nur in gut begründeten Fällen kann auf eine klinische Leistungsstudie verzichtet werden (IVDR Anhang XIII, Teil A, Absatz 1.2.3).
- Die Entwicklung von Software, die Teil des Produkts wird, muss die „Grundsätze des Software-Lebenszyklus, des Risikomanagements einschließlich der Informationssicherheit, der Verifizierung und der Validierung berücksichtigen“ (MDR Anhang I, 17.2 bzw. IVDR Anhang I, 16.2).
Derzeit gibt es keine Gesetze und harmonisierten Normen, die speziell den Einsatz des Machine Learnings in Medizinprodukten regulieren. Allerdings sind zwei Normen in Entwicklung:
- ISO 24971-2 zur Anwendung des Risikomanagements nach ISO 14971 bei KI-basierten Medizinprodukten
- IEC 62366-3 zur Anwendung des Usability Engineerings nach IEC 62366-1 bei KI-basierten Medizinprodukten. Diese Norm wird wahrscheinlich die Inhalte aus diesem Dokument übernehmen, an denen das Johner Institut ebenfalls mitgewirkt hat.
b) KI-Verordnung der EU (AI Act)
Der AI Act betrifft die allermeisten Medizinprodukte und IVD, welche Verfahren der künstlichen Intelligenz verwenden, insbesondere des Machine Learnings. Wenn bei diesen eine Benannte Stelle in die Konformitätsbewertung einbezogen werden muss (sprich: wenn die Produkte nicht in die niedrigste Klasse fallen), dann zählen diese Produkte sogar als Hochrisiko-Produkte im Sinne des AI Acts.
Ein ausführlicher Artikel zum AI Act stellt die Anforderungen dieser EU-Verordnung vor, beschreibt die Auswirkungen auf Hersteller und gibt Tipps zur Umsetzung.
Nutzen Sie auch die Übersicht über alle regulatorischen Anforderungen an Medizinprodukte.
In dieser Podcast-Episode erklärt der Experte Dr. Till Klein im Gespräch mit Prof. Johner, was das Ziel des AI Act ist, welche konkreten Anforderungen dieser stellt und unter welchen Umständen Medizinprodukte- und IVD-Hersteller betroffen sind.
Diese und weitere Podcast-Episoden finden Sie auch hier.
Die EU will ihre Digitalregulierung im Rahmen eines „digitalen Omnibus“ überarbeiten. Dazu zählt auch die KI-Regulierung. Noch sind jedoch keine konkreten Planungen bekannt.
2. Gesetzliche Anforderungen an den Einsatz von Machine Learning bei Medizinprodukten in den USA
a) Unspezifische Anforderungen
Die FDA stellt viele Anforderungen, die für Produkte mit Machine Learning unspezifisch, aber relevant sind:
- 21 CFR part 820 (u. a. part 820.30 mit den Design Controls)
- Software Validation Guidance
- Off-the-shelf-Software (OTSS) Guidance
- Cybersecurity Guidance
Die Executive Order 14110 hat die Trump-Regierung bereits am 25. Januar 2025 wieder gekippt.
Bestehen bleibt das (freiwillige) AI Risk Management Framework des NIST, einschließlich des Generative AI Profile NIST AI 600-1.
b) Spezifische Anforderungen
Frameworks
Die FDA veröffentlichte im April 2019 den Entwurf Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD). Darin führt Sie das Konzept der „Predetermined Change Control Plans“ ein.
Die Hersteller können in solch einem PCCP geplante Änderungen beschreiben und bereits mit der Zulassung des Produkts genehmigen lassen. Dann erwartet die Behörde keine Neueinreichung, wenn diese Änderungen umgesetzt werden.
- Verbesserung der klinischen und analytischen Leistungsfähigkeit: Diese ließe sich durch ein Trainieren mit mehr Datensätzen erreichen.
- Änderung der „Input-Daten“, die der Algorithmus verarbeitet. Das können zusätzliche Labordaten sein oder Daten eines anderen CT-Herstellers.
- Änderung der Zweckbestimmung: Als Beispiel nennt die FDA, dass der Algorithmus anfangs nur einen „Confidence Score“ berechnet, der die Diagnose unterstützen soll, und später die Diagnose direkt berechnet. Auch eine Änderung der vorgesehenen Patientenpopulation zählt als eine Änderung der Zweckbestimmung.
Dieser Ansatz fordert mehrere Voraussetzungen, die die FDA Säulen nennt:
- Qualitätsmanagementsystem und „Good Machine Learning Practices“ (GMLP)
- Planung und initiale Bewertung bezüglich Sicherheit und Leistungsfähigkeit
- Ansatz, um Änderungen nach der initialen Freigabe zu bewerten
- Transparenz und Überwachung der Leistungsfähigkeit im Markt
Inzwischen hat die FDA diese Anforderungen konkretisiert in ihrem Dokument „Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD)“.
Guidance Documents
Dieses Framework hat die Behörde im April 2023 in das Guidance-Dokument „Marketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence/Machine Learning (AI/ML)-Enabled Device Software Functions“ überführt.
Die FDA hat die Guidance zu den Predetermined Change Control Plans für KI-basierte Produkte aus dem Dezember 2024 im August 2025 bereits wieder überarbeitet. Die Änderungen sind allerdings überschaubar. Allerdings hat die Behörde die Definition von Machine Learning geändert.
Ein weiteres Guidance-Dokument der FDA zur radiologischen Bildgebung adressiert KI-basierte Medizinprodukte nicht direkt; es ist dennoch hilfreich. Zum einen arbeiten viele KI-/ML-basierte Medizinprodukte mit radiologischen Bilddaten, und zum anderen nennt das Dokument Fehlerquellen, die insbesondere auch für ML-basierte Produkte relevant sind:
- Patientencharakteristiken
Demografische und physiologische Charakteristiken, Bewegungsartefakte, Implantate, räumlich heterogene Verteilung des Gewebes, Verkalkungen usw. - Aufnahmecharakteristiken
Positionierung, spezifische Eigenschaften der Medizingeräte, Aufnahmeparameter (z. B. Sequenzen beim MRT oder Röntgendosen beim CT), Algorithmen zur Rekonstruktion der Bilder, externe Störquellen usw. - Bildverarbeitung
Filterung, verschiedene Software-Versionen, manuelle Selektion und Segmentierung von Bereichen, Fitting von Kurven usw.
Im Januar 2026 hat die FDA einen Entwurf des Guidance Documents „Artificial Intelligence-Enabled Device Software-Function“: Lifecycle Management and Marketing Submission Recommendations“ veröffentlicht. Sie beschreibt darin ihre Erwartungen bezüglich:
- Qualitätsmanagement
- Produktbeschreibung (z.B. Tatasche, dass und wozu KI verwendet wird; erweiterte Zweckbestimmung mit Anforderungen an Anwender, klinischen Kontext usw.; auch Beschreibung der UI-Elemente)
- User Interface (genauere Beschreibung, wie die Nutzer mit Produkt und KI inteagieren)
- Labeling (Schwerpunkt ist die Transparenz, dass KI genutzt wird, deren Limitierungen, usw. Zudem erwartete Inputs, Erklärung der Outputs, des Modells sowie der Performance)
- Risikomanagement (wenig KI-Spezifisches)
- Datenmanagement (Erklärung, wie Daten gesammelt, verarbeitet, annotiert, zum Training genutzt usw. werden. Auch Stellungnahme zu Patientenkollektiven und Biases)
- Modellbeschreibung und Modellentwicklung
- Validierung (v.a. Verweise auf Usability, auch einige Hinweise zur Validierung der Modelle selbst wie die Prüfung der Leistungsmetriken in Patientensubgruppen; zudem Vorgaben zum „Study Design“)
- Monitoring des Produkt im Markt
- Cybersecurity (auch KI-spezifische Gefährdungen wie Data Poisoning, Data Leakage ussw. Der Zusammenhang zwischen Cyberattacks einerseits und Overfitting, Model Bias und Performance Drift andererseits wird nicht ganz klar.)
Aus diesem Dokument ergeben sich auch Anforderungen an die Dokumentation, die die FDA bei einer Einreichung erwartet.
Weitere Best Practices
Die FDA, Health Canada und die britische MHRA (Medicines and Healthcare Products Regulatory Agency) haben in Zusammenarbeit die Good Machine Learning Practice for Medical Device Development: Guiding Principles veröffentlicht. Das Dokument enthält zehn Leitprinzipien, die man beim Einsatz von maschinellem Lernen in Medizinprodukten beachten sollte. Aufgrund der Kürze von nur zwei Seiten geht das Dokument nicht ins Detail, bringt aber die wichtigsten Prinzipien auf den Punkt.
Ergänzung
Wertvoll sind die aktualisierten „Guiding Principles“ für Medizinprodukte, welche Verfahren des maschinellen Lernens verwenden. Diese hat die FDA gemeinsam mit Health Canada entwickelt. Damit gibt es zwei Listen:
- Good Manufacturing Learning Practices: Guiding Principles
- Transparency for Machine Learning-Enabled Medical Devices: Guiding Principles
Das zweite und neue Dokument ist das speziellere, das die Prinzipien 7 und 9 aus dem ersten Dokument genauer ausführt.
c) Anforderungen einzelner Staaten
Die KI-Regulierung in den USA ist nicht homogen. Die einzelnen Bundesstaaten haben individuelle Vorgaben:
- Kalifornien:
- Colorado: Artificial Intelligence Act (v. a. KI-Verbraucherschutz)
3. Gesetzliche Anforderungen an den Einsatz von Machine Learning bei Medizinprodukten in anderen Ländern
a) China: NMPA
Die chinesische NMPA hat den Entwurf „Technical Guiding Principles of Real-World Data for Clinical Evaluation of Medical Devices“ zur Kommentierung freigegeben.
Dieses Dokument ist derzeit aber nur auf Chinesisch verfügbar. Wir haben das Inhaltsverzeichnis automatisiert übersetzen lassen. Das Dokument adressiert:
- Anforderungsanalyse
- Datensammlung und -aufbereitung
- Entwurf des Modells
- Verifizierung und Validierung (auch klinische Validierung)
- Post-Market Surveillance
Die Behörde rüstet personell auf und hat eine „AI Medical Device Standardization Unit“ gegründet. Diese kümmert sich um die Standardisierung von Terminologien, Technologien und Prozessen für die Entwicklung und Qualitätssicherung.
Seit dem 01.09.25 gibt es zudem Vorgaben zu Inhalten generierter KI und zum Labeling sowie zu Registrierungspflichten. Der Fokus sind die Inhalte(!) von generierter KI.
b) Japan
Das japanische Ministry of Health, Labour and Welfare arbeitet ebenfalls an AI-Standards. Den Fortschritt dieser Bemühungen veröffentlicht die Behörde leider nur auf Japanisch. (Übersetzungsprogramme helfen aber weiter.) Konkrete Ergebnisse stehen derzeit noch aus.
Zudem hat Japan den „AI Promotion Act“ veröffentlicht, dessen Ziel der Name offenbart. Es geht also nicht primär um ein Verbot oder Begrenzung der KI-Nutzung.
c) Weitere Länder
- Schweiz: Der Bundesrat unterzeichnete am 12.02.25 die KI-Rahmenkonvention des Europarats (CAI).
- Kanada: Hier ist der Artificial Intelligence and Data Act (AIDA) als Teil von Bill C-27 zu beachten, der allerdings noch in Entwicklung ist.
- Australien: Bereits in einer zweiten Version verfügbar sind die Voluntary AI Safety Standards.
4. Für das Machine Learning relevante Normen und Best Practices
a) Best Practices
Die folgende Tabelle verschafft einen Überblick:
| Herausgeber | Titel | Inhalte | Bewertung |
| COICR | Artificial Intelligence in Healthcare | Keine konkreten neuen Anforderungen; verweist auf bestehende und empfiehlt die Entwicklung von Normen | wenig hilfreich |
| Xavier University | Perspectives and Good Practices for AI and Continuously Learning Systems in Healthcare. | Es geht (auch) um kontinuierlich lernende Systeme. Dennoch lassen sich viele der genannten Best Practices auch auf nicht kontinuierlich lernende Systeme übertragen. | Hilfreich insbesondere bei kontinuierlich lernenden Systemen. Der Leitfaden des Johner Instituts ist berücksichtigt. |
| Das „International Software Testing Qualification Board“ | Certified Tester AI Testing (CT-AI) Syllabus | ISTQB stellt einen Lehrplan zum Testen von KI-Systemen zum Download bereit. | empfehlenswert |
| ANSI gemeinsam mit CSA (Consumer Technology Association) | Definitions and Characteristics of Artificial Intelligence (ANSI/CTA-2089) Definitions/Characteristics of Artificial Intelligence in Health Care (ANSI/CTA-2089.1) | Definitionen | nur als Sammlung von Definitionen hilfreich |
| WHO/ITU | Good practices for health applications of machine learning: Considerations for manufacturers and regulators | Das Johner Institut hat wesentliche Inputs für diese AI4H-Initiative eingebracht. Eine Abstimmung dieser Ergebnisse mit dem IMDRF ist geplant. | Empfehlenswert, aber bereits im Leitfaden des Johner Instituts berücksichtigt |
| TeamNB und IG-NB | Questionnaire des IG_NB Questionnaire des TeamNB | Beide basieren auf dem Leitfaden des Johner Instituts. | „Must read“, da von Benannten Stellen verwendet |
| IMDRF | Good machine learning practice for medical device development – Guiding Principles |
b) Normen
Einzelne Normen
| Titel | Inhalte | Bewertung |
| IEC/TR 60601-4-1 | Vorgaben für „Medizinische elektrische Geräte und medizinische elektrische Systeme mit einem Maß an Autonomie“. Diese Vorgaben sind allerdings nicht spezifisch für Medizinprodukte, die Verfahren des Machine Learnings verwenden. | bedingt hilfreich |
| SPEC 92001-1 „Künstliche Intelligenz – Life Cycle Prozesse und Qualitätsanforderungen – Teil 1: Qualitäts-Meta-Modell“ | Stellt ein Meta-Modell vor, nennt aber keine konkreten Anforderungen an die Entwicklung von AI/ML-Systemen. Das Dokument ist unspezifisch und nicht auf eine bestimmte Branche ausgerichtet. | wenig hilfreich |
| SPEC 92001-2 „Künstliche Intelligenz – Life-Cycle-Prozesse und Qualitätsanforderungen – Teil 2: Robustheit“ | Enthält im Gegensatz zum ersten Teil konkrete Anforderungen. Diese zielen v. a. auf das Risikomanagement. Sie sind jedoch unspezifisch für Medizinprodukte. | |
| ISO/IEC CD TR 29119-11 | Wir haben diese Norm für Sie gelesen und bewertet. | ignorieren |
| ISO 24028 – Overview of Trustworthiness in AI | Unspezifisch für eine bestimmte Domäne, nennt aber auch Beispiele für das Gesundheitswesen. Keine konkreten Empfehlungen und keine spezifischen Anforderungen. Mindmap mit Kapitelübersicht | bedingt empfehlenswert |
| ISO 23053 – Framework for AI using Machine Learning | Leitfaden für einen Entwicklungsprozess von ML-Modellen. Er enthält keine konkreten Anforderungen, stellt aber den Stand der Technik dar. | bedingt empfehlenswert |
| ISO/IEC 25059:2023 Software engineering — Systems and software Quality Requirements and Evaluation (SQuaRE) — Quality model for AI systems | Noch nicht analysiert | |
| ISO/DTS 24971-2 Medical devices — Guidance on the application of ISO 14971 Part 2: Machine learning in artificial intelligence | Liegt als Draft vor. Konkreter Fokus: „Machine Learning in Artificial Intelligence“ (sic!). Wieder ist Pat Baird federführend, wie bei der BS 34971, einer Leitlinie zum gleichen Thema. | empfehlenswert insbesondere als Checkliste, auch wenn Konzepte inkonsistent sind |
| BS/AAMI 34971 | vergleichbar ISO/TS 24971-2 Mindmap mit Kapitelstruktur | besser ISO/TS 24971-2 lesen; sehr teuer |
| BS ISO/IEC 23894:2023 „Information technology. Artificial intelligence. Guidance on risk management”. | Keine Dopplung zur BS 39471, denn diese Norm ist nicht spezifisch für Medizinprodukte. | ignorieren |
| BS 30440:2023 „Validation framework for the use of artificial intelligence (AI) within healthcare. Specification”. | Diese Norm sieht nicht nur Hersteller, sondern auch Betreiber, Krankenversicherungen und Anwender als Lesende. | |
| ISO/IEC 42001:2023 „Information technology – Artificial intelligence – Management system“. | Bitte diesen Fachartikel zur ISO/IEC 42001 beachten, der eine Übersicht über die Norm und deren Anforderungen sowie konkrete Praxistipps zur Implementierung gibt. | in einigen Märkten verpflichtend |
| ISO/IEC 5338:2023 Information technology — Artificial intelligence — AI system life cycle processes | Beschreibt insgesamt 33 Prozesse; kein Bezug zu Medizinprodukten. Starke Orientierung an Software-Entwicklung; referenziert durchgängig die Software-Normen ISO/IEC/IEEE12207 und ISO/IEC/IEEE 15288 | bedingt empfehlenswert |
| ISO/IEC TS 4213:2022 Information technology — Artificial intelligence — Assessment of machine learning classification performance | Die Norm listet statistische Verfahren, abhängig von der Aufgabenstellung des Modells. Sie nennt für binäre Klassifikationen u. a. die 4-Felder-Tafel („confusion matrix“), den F1-Score und die „Lift curve“. Für die „Multi-label classification“ nennt sie „Hamming loss“, „Jaccard index“ und weitere. | Die Verfahren sind so knapp beschrieben, dass der Wert der Norm eher darin besteht, für die Aufgabenstellung des Modells die passenden Verfahren für die Leistungsmessung zusammenzutragen. Der Erkenntnisgewinn für erfahrene Data Scientists und Statistiker ist daher unklar. Einen Teil des Werts liefert das Inhaltsverzeichnis der ISO/IEC 4214:2022. |
Die Videotrainings im Auditgarant stellen wichtige Verfahren wie LRP LIME, die Visualisierung der Aktivierung von neuronalen Schichten oder Counterfactuals vor.
Normen der IEEE
Eine ganze Familie an Normen ist bei der IEEE in Entwicklung:
- P7001 – Transparency of Autonomous Systems
- P7002 – Data Privacy Process
- P7003 – Algorithmic Bias Considerations
- P7009 – Standard for Fail-Safe Design of Autonomous and Semi-Autonomous Systems
- P7010 – Wellbeing Metrics Standard for Ethical Artificial Intelligence and Autonomous Systems
- P7011 – Standard for the Process of Identifying and Rating the Trustworthiness of News Sources
- P7014 – Standard for Ethical Considerations in Emulated Empathy in Autonomous and Intelligent Systems
- 1 – Standard for Human Augmentation: Taxonomy and Definitions
- 2 – Standard for Human Augmentation: Privacy and Security
- 3 – Standard for Human Augmentation: Identity
- 4 – Standard for Human Augmentation: Methodologies and Processes for Ethical Considerations
- P2801 – Recommended Practice for the Quality Management of Datasets for Medical Artificial Intelligence
- P2802 – Standard for the Performance and Safety Evaluation of Artificial Intelligence Based Medical Device: Terminology
- P2817 – Guide for Verification of Autonomous Systems
- 1.3 – Standard for the Deep Learning-Based Assessment of Visual Experience Based on Human Factors
- 1 – Guide for Architectural Framework and Application of Federated Machine Learning
ISO-Normen, die in Entwicklung sind
Auch bei der ISO arbeiten mehrere Arbeitsgruppen an AI/ML-spezifischen Normen:
- ISO 20546 – Big Data – Overview and Vocabulary
- ISO 20547-1 – Big Data reference architecture – Part 1: Framework and application process
- ISO 20547-2 – Big Data reference architecture – Part 2: Use cases and derived requirements
- ISO 20547-3 – Big Data reference architecture – Part 3: Reference architecture
- ISO 20547-5 – Big Data reference architecture – Part 5: Standards roadmap
- ISO 22989 – AI Concepts and Terminology
- ISO 24027 – Bias in AI systems and AI aided decision making
- ISO 24029-1 – Assessment of the robustness of neural networks – Part 1 Overview
- ISO 24029-2 – Formal methods methodology
- ISO 24030 – Use cases and application
- ISO 24368 – Overview of ethical and societal concerns
- ISO 24372 – Overview of computations approaches for AI systems
- ISO 24668 – Process management framework for Big data analytics
- ISO 38507 – Governance implications of the use of AI by organizations.
Normenfamilie ISO/IEC 5259
Ebenfalls eine ganze Normenfamilie beschäftigt sich mit „Artificial intelligence — Data quality for analytics and machine learning (ML)“.
| Norm | Titel |
| ISO/IEC 5259-1 | Part 1: Overview, terminology, and examples |
| ISO/IEC 5259-2 | Part 2: Data quality measures |
| ISO/IEC 5259-3 | Part 3: Data quality management requirements and guidelines |
| ISO/IEC 5259-4 | Part 4: Data quality process framework |
| ISO/IEC 5259-5 | Part 5: Data quality governance framework |
5. Tipps zum Erfüllen der gesetzlichen Anforderungen
Tipp 1: Explainability nutzen
Allgemeines
Mit der Aussage, dass die Verfahren des Machine Learnings Blackboxes darstellen würden, sollten sich Auditoren nicht mehr pauschal zufriedengeben.
Es gibt vielversprechende Ansätze in der aktuellen Forschungsliteratur, wie die Vorhersagen von Deep-Learning-Modellen plausibilisiert werden können. Zum Beispiel kann man bei der Klassifikation von Bildern nachvollziehen, welche Input-Pixel für die Klassifikation entscheidend sind (s. u.).
Es haben sich jedoch noch keine Standard-Methoden etabliert, da die derzeitigen Verfahren unterschiedliche Stärken und Schwächen haben und sich der Status quo in einer heuristischen Phase befindet. Es ist jedoch davon auszugehen, dass die Forschung in diesem Bereich in den nächsten Jahren weitere Fortschritte Richtung Erklärbarkeit machen wird.
Viele Ansätze richten sich derzeit „nur“ auf die Erklärung von konkreten Einzelvorhersagen anhand der Eingabedaten (lokale Erklärbarkeit).
Beispiel
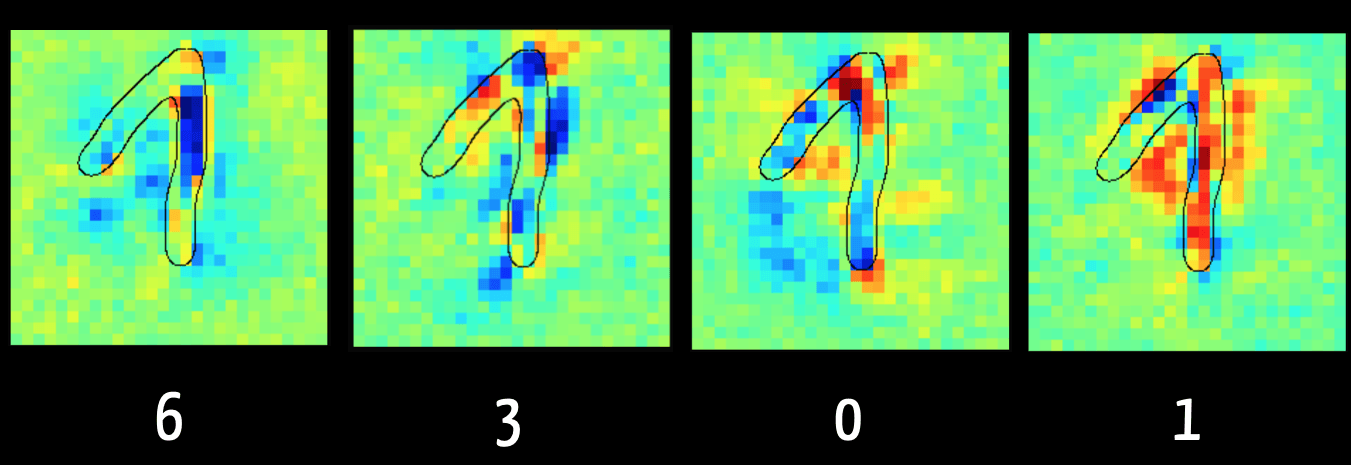
Mit der Layer Wise Relevance Propagation kann man bei einigen Modellen erkennen, welche Input-Daten („Feature“) für den Algorithmus entscheidend waren, z. B. die Klassifikation.
Abbildung 1 zeigt im linken Bild, dass der Algorithmus die Ziffer „6“ vor allem wegen der dunkelblau markierten Pixel ausschließen kann. Das ergibt Sinn, weil bei einer „6“ dieser Bereich typischerweise keine Pixel erhält. Hingegen sind im rechten Bild die Pixel rot, die den Algorithmus in der Annahme bestärken, dass die Ziffer eine „1“ ist.
Der Algorithmus bewertet die Pixel im aufsteigenden Schenkel der Ziffer eher als schädlich für die Klassifizierung als „1“. Das liegt daran, dass er mit Bildern trainiert wurde, bei denen die „1“ nur als ein senkrechter Strich geschrieben wird, wie dies in den USA der Fall ist. Dies verdeutlicht, wie relevant es für das Ergebnis ist, dass die Trainingsdaten repräsentativ für die später zu klassifizierenden Daten sind.
Besonders lesenswert ist das online kostenfrei verfügbare Buch „Interpretable Machine Learning“ von Christoph Molnar, einem der Keynote Speaker beim Institutstag 2019.
Tipp 2: Den Stand der Technik regelmäßig bestimmen
Hersteller sind gut beraten, die Fragen mancher Auditoren nach dem Stand der Technik nicht global zu beantworten, sondern zu unterscheiden:
- Technische Umsetzung: Einschlägige Standards wie die hier genannten helfen beim Nachweis, dass die Entwicklung und Verifizierung bzw. Validierung der Software und der Modelle den aktuellen Best Practices entspricht.
- Leistungsparameter: Die Hersteller sollten die Leistungsfähigkeit mit klassischen Verfahren sowie anderen Modellen und Algorithmen des Machine Learnings vergleichen. Dieser Vergleich sollte anhand aller relevanten Attribute erfolgen, wie Sensitivität, Spezifität, Robustheit, Performanz, Wiederholbarkeit, Erklärbarkeit und Akzeptanz.
Tipp 3: Mit dem KI-Leitfaden arbeiten
Der Leitfaden zur Anwendung der künstlichen Intelligenz (KI) bei Medizinprodukten steht jetzt kostenfrei bei GitHub zur Verfügung.
Diesen Leitfaden haben wir gemeinsam mit Benannten Stellen, Herstellern und KI-Experten entwickelt.
- Er hilft Herstellern, KI-basierte Produkte gesetzeskonform zu entwickeln und schnell und sicher in den Markt zu bringen.
- Interne und externe Auditoren sowie Benannte Stellen nutzen den Leitfaden, um die Gesetzeskonformität KI-basierter Medizinprodukte und den zugehörigen Lebenszyklusprozess zu prüfen.
Nutzen Sie die Excel-Version des Leitfadens, die hier kostenlos erhältlich ist. Damit können Sie die Anforderungen des Leitfadens filtern, in eigene Vorgabedokumente übernehmen und an Ihre spezifische Situation anpassen.
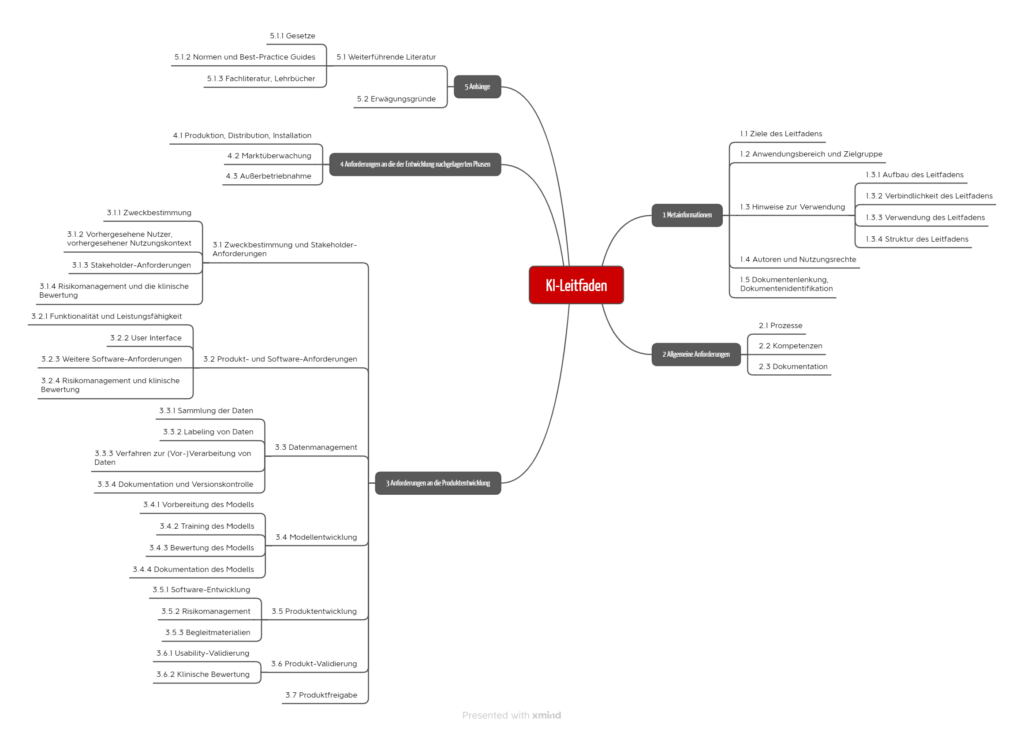
Beim Schreiben war es uns wichtig, den Herstellern und Benannten Stellen präzise Prüfkriterien an die Hand zu geben, die eine eindeutige und unstrittige Bewertung erlauben. Weiterhin steht der Prozessansatz im Vordergrund. Entlang dieser Prozesse sind die Anforderungen des Leitfadens gruppiert (s. Abb. 3).
Tipp 4: Sich auf die typischen Fragen im Audit vorbereiten
Allgemeines
Noch haben sich die Benannten Stellen und Behörden nicht auf ein einheitliches Vorgehen und auf gemeinsame Anforderungen bei Medizinprodukten mit maschinellem Lernen geeinigt.
Daher tun sich Hersteller regelmäßig schwer mit dem Nachweis, dass die an das Produkt gestellten Anforderungen an etwa Genauigkeit, Korrektheit und Robustheit erfüllt sind.
Dr. Rich Caruana, einer der führenden Köpfe bei Microsoft im Bereich der künstlichen Intelligenz, riet sogar vom Einsatz eines von ihm selbst entwickelten neuronalen Netzwerks ab, das Patienten mit Lungenentzündung die passende Therapie vorschlagen sollte:
„I said no. I said we don’t understand what it does inside. I said I was afraid.”
Dr. Rich Caruana, Microsoft
Dass es Maschinen gibt, die ein Anwender nicht versteht, ist nicht neu. Man kann eine PCR anwenden, ohne sie zu verstehen; es gibt auf jeden Fall Menschen, die die Funktionsweise und das Innenleben dieses Produkts kennen. Bei der künstlichen Intelligenz ist das jedoch nicht mehr gegeben.
Leitfragen
Zu den Fragen, die Auditoren Herstellern von Produkten mit Machine Learning stellen sollten, zählen beispielsweise:
| Leitfrage | Hintergrund |
| Weshalb glauben Sie, dass Ihr Produkt dem Stand der Technik entspricht? | Klassische Einstiegsfrage. Hier sollten Sie auf technische und medizinische Aspekte eingehen. |
| Wie kommen Sie zur Annahme, dass Ihre Trainingsdaten keinen Bias haben? | Andernfalls wären die Ergebnisse falsch bzw. nur unter bestimmten Voraussetzungen richtig. |
| Wie haben Sie ein Overfitting Ihres Modells vermieden? | Sonst würde der Algorithmus nur die Daten richtig vorhersagen, mit denen er trainiert wurde. |
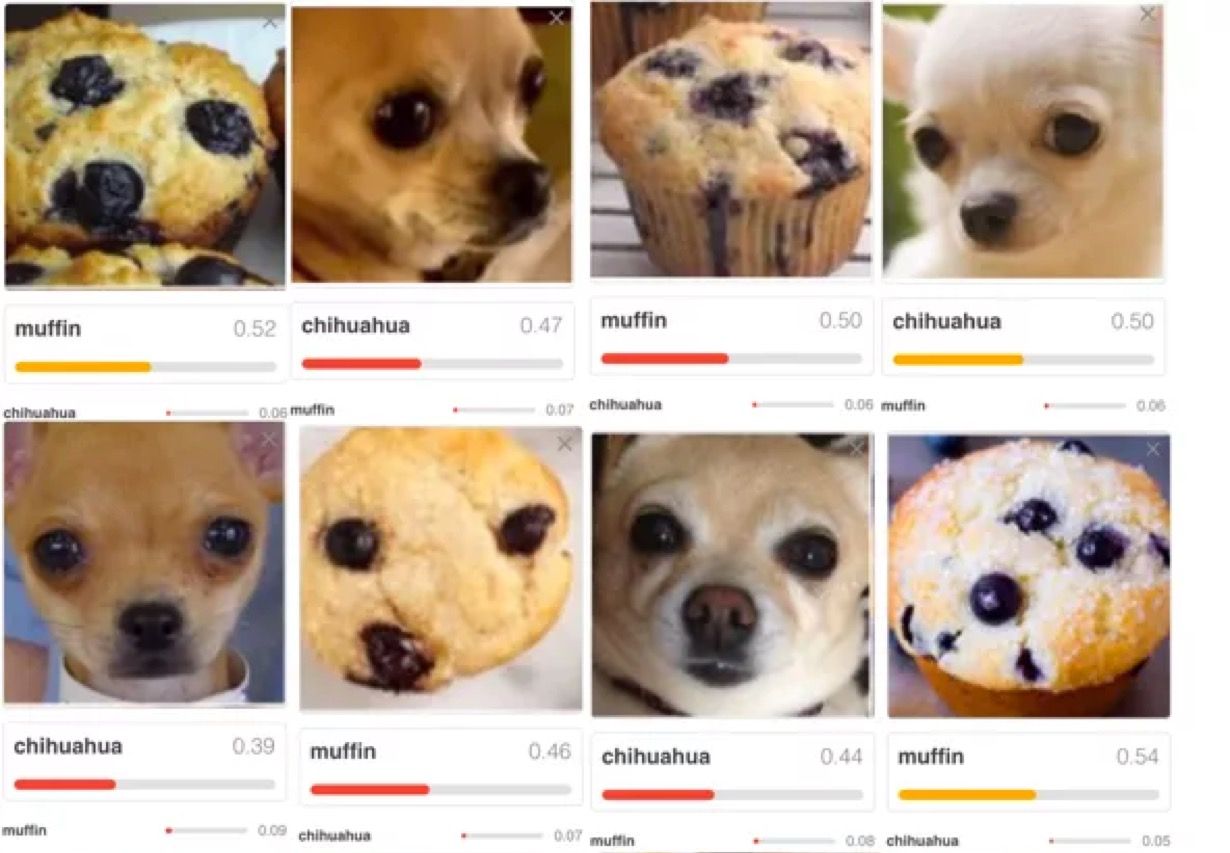
| Was veranlasst Sie zur Annahme, dass die Ergebnisse nicht nur zufällig richtig sind? | Es könnte sein, dass ein Algorithmus korrekt entscheidet, dass auf einem Bild ein Haus zu erkennen ist. Der Algorithmus hat aber kein Haus, sondern den Himmel erkannt. Ein weiteres Beispiel zeigt die Abb. 4. |
| Welche Voraussetzungen müssen Daten erfüllen, damit Ihr System sie richtig klassifiziert bzw. die Ergebnisse richtig vorhersagt? Welche Randbedingungen sind einzuhalten? | Da das Modell mit einer bestimmten Menge an Daten trainiert wurde, kann es nur für Daten, die aus der gleichen Grundgesamtheit stammen, korrekte Vorhersagen treffen. |
| Wären Sie mit einem anderen Modell oder mit anderen Hyperparametern zu einem besseren Ergebnis gekommen? | Hersteller müssen Risiken weitestgehend minimieren. Dazu zählen auch Risiken durch falsche Vorhersagen suboptimaler Modelle. |
| Weshalb gehen Sie davon aus, dass Sie ausreichend viele Trainingsdaten verwendet haben? | Das Sammeln, Aufbereiten und „Labeln“ von Trainingsdaten ist aufwendig. Je größer die Datenmenge ist, mit der ein Modell trainiert wird, desto leistungsfähiger kann es sein. |
| Welchen Standard haben Sie beim Labeling der Trainingsdaten verwendet? Weshalb betrachten Sie den gewählten Standard als Gold-Standard? | Besonders wenn die Maschine beginnt, den Menschen überlegen zu sein, wird es schwierig, festzulegen, ob ein Arzt, eine Gruppe von „normalen“ Ärzten oder die weltweit besten Experten einer Fachrichtung die Referenz sind. |
| Wie können Sie die Reproduzierbarkeit gewährleisten, wenn Ihr System weiter lernt? | Besonders bei Continuously Learning Systems (CLS) muss gewährleistet bleiben, dass durch das weitere Training die Leistungsfähigkeit zumindest nicht abnimmt. |
| Haben Sie Systeme validiert, die Sie zum Sammeln, Vorbereiten und Analysieren der Daten sowie zum Trainieren und Validieren Ihrer Modelle verwenden? | Ein wesentlicher Teil der Arbeit besteht darin, die Trainingsdaten zu sammeln und aufzubereiten sowie das Modell damit zu trainieren. Die dazu notwendige Software ist nicht Teil des Medizinprodukts. Sie unterliegt aber den Anforderungen an die Computerized Systems Validation. |
Hintergrund zu den Leitfragen
Die o. g. Fragen sind typischerweise auch im Rahmen des Risikomanagements nach ISO 14971 und der klinischen Bewertung gemäß MEDDEV 2.7.1 Revision 4 (bzw. Leistungsbewertung von IVD) zu erörtern.
Hinweise, wie Hersteller diese regulatorischen Anforderungen an Medizinprodukte mit Machine Learning erfüllen können, gibt der Artikel Künstliche Intelligenz in der Medizin. Beachten Sie auch den Beitrag Wie sich klinische Studien bei Medizinprodukten mit künstlicher Intelligenz vermeiden lassen sowie den Kategorieartikel zu Regulatory Affairs.
Tipp 5: Unterstützung nutzen
Entwickeln Sie Medizinprodukte, die künstliche Intelligenz nutzen? Das Johner Institut unterstützt Hersteller dabei,
- Produkte gesetzeskonform zu entwickeln und in Verkehr zu bringen,
- Verifizierungs- und Validierungsaktivitäten zu planen und durchzuführen,
- Produkte auf Nutzen, Leistungsfähigkeit und Sicherheit zu bewerten,
- die Eignung der Verfahren (insbesondere der Modelle) und der Trainingsdaten zu bewerten,
- die regulatorischen Anforderungen an die Post-Market-Phase zu erfüllen und
- passgenaue Verfahrensanweisungen zu erstellen.
Sie finden hier eine vollständigere Übersicht.
Tipp 6: Typische Fehler von KI-Startups vermeiden
Viele Startups, die Verfahren der künstlichen Intelligenz, insbesondere des Machine Learning nutzen, beginnen die Produktentwicklung mit den Daten. Dabei unterlaufen ihnen häufig die gleichen Fehler:
| Fehler | Folgen |
| Die Software und die Prozesse zum Sammeln und Aufbereiten der Trainingsdaten sind nicht validiert. Regulatorische Anforderungen sind bestenfalls in Ansätzen bekannt. | Im schlimmsten Fall können die Daten und Modelle nicht genutzt werden. Das wirft die ganze Entwicklung wieder auf den Anfang zurück. |
| Die erklärte Performance der Produkte leiten die Hersteller nicht aus der Zweckbestimmung und dem Stand der Technik ab, sondern aus der Leistungsfähigkeit der Modelle. | Die Produkte scheitern in der klinischen Bewertung. |
| Menschen, deren wirkliche Leidenschaft die Data Science oder die Medizin ist, versuchen sich als Unternehmensentwickler. | Die Produkte schaffen es nie bis auf den Markt oder treffen den tatsächlichen Bedarf nicht. |
| Das Geschäftsmodell bleibt zu lange zu vage. | Die Investoren halten sich zurück oder/und das Unternehmen trocknet finanziell aus und scheitert. |
Startups können sich bei uns melden. In wenigen Stunden können wir helfen, diese fatalen Fehler zu vermeiden.
Die Direktorin „Global Regulatory Affairs“, Carmen Bellebna, berichtet in dieser Episode darüber, wie sie und ihr Unternehmen DeepEye die Anforderungen des AI Act erfüllen.
Diese und weitere Podcast-Episoden finden Sie auch hier.
6. Fazit und Zusammenfassung
a) Regulatorische Anforderungen
Die regulatorischen Anforderungen sind eindeutig. Doch Herstellern und teilweise auch Behörden und Benannten Stellen bleibt unklar, wie diese für Medizinprodukte, die Verfahren des Machine Learnings nutzen, zu interpretieren und konkret umzusetzen sind.
b) Zu viele und nur bedingt hilfreiche Best Practice Guides
Daher fühlen sich viele Institutionen berufen, mit „Best Practices“ zu helfen. Leider sind viele dieser Dokumente nur bedingt hilfreich:
- Sie wiederholen Lehrbuchwissen über die künstliche Intelligenz im Allgemeinen und das Machine Learning im Speziellen.
- Die Guidance-Dokumente ergehen sich in Selbstverständlichkeiten und Banalitäten.
Wer nicht bereits vor dem Lesen dieser Dokumente wusste, dass das Machine Learning zu Fehlklassifizierungen und Bias führen und damit Patienten gefährden oder benachteiligen kann, sollte keine Medizinprodukte entwickeln. - Viele dieser Dokumente beschränken sich darauf, die für das Machine Learning spezifischen Probleme aufzulisten, die die Hersteller adressieren müssen. Es fehlen Best Practices, wie man diese Probleme minimiert.
- Wenn es Empfehlungen gibt, sind diese meist wenig konkret. Sie bieten keine ausreichende Handlungsleitung.
- Es dürfte den Herstellern und Behörden schwerfallen, aus Textwüsten wirklich prüfbare Anforderungen zu extrahieren.
Leider scheint keine Besserung in Sicht zu sein, im Gegenteil: Es werden immer mehr Richtlinien entwickelt. Beispielsweise empfiehlt die OECD die Entwicklung von AI/ML-spezifischen Standards und erarbeitet derzeit selbst einen. Gleiches gilt für die IEEE und das DIN und viele weitere Organisationen.
Fazit:
- Es gibt zu viele Normen, um den Überblick behalten zu können. Und es werden kontinuierlich mehr.
- Die Normen überlappen stark und sind überwiegend von eingeschränktem Nutzen. Sie enthalten keine (binär entscheidbaren) Prüfkriterien.
- Sie kommen (zu) spät.
c) Qualität statt Quantität
Hersteller von Medizinprodukten benötigen bei den Best Practices und Normen zum Machine Learning mehr Qualität und nicht mehr Quantität.
Best Practices und Normen sollten handlungsleitend sein und überprüfbare Anforderungen stellen. Dass die WHO den Leitfaden des Johner Instituts aufgreift, gibt Anlass zu vorsichtigem Optimismus.
Es wäre wünschenswert, wenn sich die Benannten Stellen, die Behörden und ggf. auch die MDCG aktiver in die (Weiter-)Entwicklung dieser Standards einbringen würden. Dies sollte in transparenter Weise geschehen. Zu welch bescheidenen Ergebnissen das Arbeiten in Hinterzimmern ohne (externe) Qualitätssicherung führt, haben wir in letzter Zeit mehrfach erfahren.
Mit einem gemeinsamen Vorgehen gelänge es, ein gemeinsames Verständnis davon zu erreichen, wie Medizinprodukte, die maschinelles Lernen verwenden, entwickelt und geprüft werden müssten. Es gäbe nur Gewinner.
Benannte Stellen und Behörden sind herzlich eingeladen, an der Weiterentwicklung der Leitfäden mitzuwirken. Eine E-Mail an das Johner Institut genügt.
Hersteller, die Unterstützung bei der Entwicklung und Zulassung ML-basierter Produkte (z. B. bei der Überprüfung der technischen Dokumentation oder bei der Validierung von ML-Bibliotheken) wünschen, können sich gerne via E-Mail oder über das Kontaktformular melden.
Änderungshistorie
2026
- 2026-01-29: Im Kapitel 2.b) einen größeren Absatz zur neuen FDA Guidance ergänzt
2025
- 2025-12-27
- Kapitel 2. a): Aufhebung der Executive Order und Hinweise zu Vorgaben der NIST ergänzt
- Kapitel 2. c) “Anforderungen einzelner Staaten“ eingefügt
- Kapitel 3. a) letzten Satz ergänzt mit Registrierungspflichten
- Kapitel 3. c) „Weitere Länder“ eingefügt
- 2025-11-28: Im Abschnitt 1.a) Hinweise auf ISO 24971-2 und IEC 62366-1 eingefügt
- 2025-11-13: Im Abschnitt 1.b) Hinweis auf „digitalen Omnibus“ ergänzt
- 2025-10-22: Umfangreiche Änderungen
- Einleitung neu geschrieben
- Key Takeaways eingefügt
- Hinweis auf Schlagwortartikel eingefügt
- Kapitel 1.a): Überschrift umbenannt, einleitenden Satz geändert, Hinweis mit fehlenden Normen als Warnung formuliert
- Kapitel 1.c) (sonstige Normen) gelöscht
- Kapitel 2.a) umformuliert und gekürzt
- Kapitel 4 (Normen, Best Practices) komplett neu geschrieben. Dabei aktualisiert und deutlich verkürzt
- Tabellen neu nummeriert
- 2025-09-04: In Abschnitt 2.c) die neue FDA Guidance zu den Predetermined Change Control Plans erwähnt
- 2025-05-05: Hinweis auf Podcast hinzugefügt
- 2025-04-14:
- Inhalte von Kapitel 4.q) in Kapitel 4.w) verschoben, da beide das IMDRF betreffen
- Das Kapitel 4.q) völlig neu geschrieben. Es behandelt jetzt die Normenfamilie ISO/IEC 5259
- In Kapitel 4.r) den Hinweis auf die neue ISO 24971-2 ergänzt
- In Kapitel 4.x) die angekündigte Bewertung der Norm ISO/IEC 5338:2023 ergänzt
- Kapitel 4. z) zur ISO/IEC 4213:2022 hinzugefügt
- 2025-02-22: Kapitel 2.b) (AI Act) komplett „entkernt“ und die Inhalte in den Artikel zum AI Act verschoben und dort umstrukturiert und aktualisiert. Verweis in Abschnitt 4.v) auf Artikel zur ISO/IEC 42001 ergänzt. Abschnitte 4.x) und 4.y) eingefügt. Link auf TeamNB in Abschnitt 4.p) ergänzt
- 2025-01-16: Abschnitt 5 neu eingefügt und strukturiert
2024
- 2024-07-08: In Abschnitt 2.c) die neue Leitlinie der FDA ergänzt, im Abschnitt 4.w) die Leitlinie des IMDRF
- 2024-03-26: Link zum verabschiedeten AI Act eingefügt.
- 2024-01-26: Hinweis zum Kompromissvorschlag zum AI Act ergänzt
- 2024-01-18: Kapitel 4.v) zur ISO 42001 eingefügt
2023
- 2023-11-20: Neues Kapitel 4.s) mit ISO/NP TS 23918 eingefügt
- 2023-11-03: Kapitelstruktur überarbeitet. Anforderungen der FDA ergänzt. Neue BS-Normen erwähnt.
- 2023-10-09: Kapitel 2.n) eingefügt, das die ISO 23053 bewertet.
- 2023-09-07: Kapitel 2.s) einfügt, das die AAMI 34971:2023-05-30 bewertet.
- 2023-07-03: Link auf BS/AAMI 34971:2023-05-30 eingefügt
- 2023-06-27: Abschnitt 1.b) i) ergänzt: AI act gilt für MP/IVD höherer Risikoklassen.
- 2023-06-23: FDA Guidance Document vom April 2023 ergänzt
- 2023-06-08: Kapitel 4 eingefügt
- 2023-02-14: Abschnitt 2.r) eingefügt
2022 und früher
- 2022-11-17: Link zum neuen Entwurf des AI Acts ergänzt
- 2022-10-21: Link zu Updates der EU zum AI Act ergänzt
- 2022-06-27: Im Kapitel zur FDA deren Guidance-Dokument für die radiologische Bildgebung und die dort genannten Fehlerquellen eingefügt
- 2021-11-01: Kapitel mit FDAs Guiding Principles eingefügt
- 2021-09-10: Link mit der Stellungnahme für die EU ergänzt
- 2021-07-31: Im Abschnitt 1.b.ii) in der Tabelle zwei Zeilen mit weiteren Kritikpunkten angehängt. In der Zeile mit den Definitionen je einen Abschnitt ergänzt.
- 2021-07-26: Abschnitt 2.n) in Abschnitt 1.b) verschoben. Dort Kritik und Handlungsaufruf ergänzt.
- 2021-04-27: Abschnitt zu Plänen der EU zu neuer KI-Regulierung ergänzt


Herzlichen Dank für den informativen und aufschlussreichen Artikel. Die detaillierte Aufschlüsselung der Herausforderungen und erforderlichen Konformitätsnachweise als auch die Sammlung der verschiedenen Normen ist besonders wertvoll.