
Unter Software-Wartung versteht man die Phase, in der die Software weiterentwickelt wird, z. B. mit dem Ziel
- Fehler zu beheben,
- neue Funktionalitäten zu implementieren oder
- die Software an eine geänderte Laufzeitumgebung anzupassen.
79% aller Bugs werden laut FDA während der Software-Wartung eingeführt. Entsprechend adressieren einige Regularien dieses Thema.
Update: Ergänzung zur Software-Wartung während der Entwicklung
Regulatorische Anforderungen an die Software-Wartung
Forderungen der Medizinprodukteverordnung MDR (2017/745)
Die Medizinprodukteverordnung MDR verlangt, dass Medizinproduktehersteller eine technische Dokumentation erstellen und abhängig von der Klasse (I, IIa, IIb, III) diese eine Benannten Stelle einreichen.
Bei einer Änderung der Software müssen Sie die technische Dokumentation aktualisieren, wobei Sie die Forderungen der ISO 13485 nach Dokumentenlenkung erfüllen sollten. Lesen Sie weiter unten, ab wann eine neue Einreichung von der Benannten Stelle erwartet wird.
Wenn die Änderung die Klasse des Produkts ändert, kann es sogar notwendig sein, dass Sie ein anderes Konformitätsbewertungsverfahren durchlaufen.
Forderungen der IEC 62304
Die IEC 62304 beschreibt einen expliziten Prozess für die Software-Wartung. Beachten Sie, dass auch andere in der Norm beschriebenen Prozesse den Ablauf einer „Software-Änderungen“ regulieren:
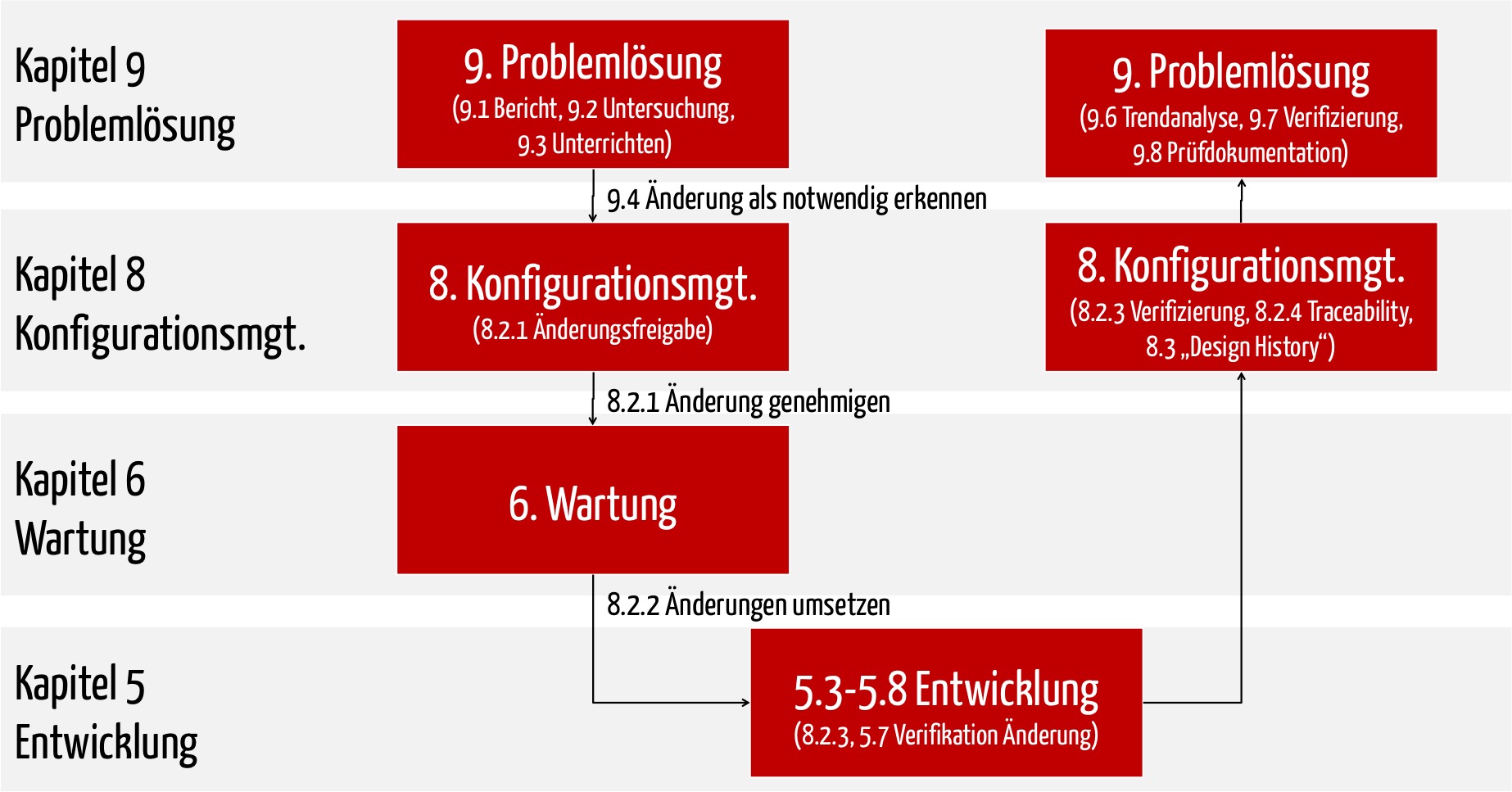
Bei Änderungen müssen Sie den Problemlösungsprozess (IEC 62304 Kapitel 9), den Konfigurationsmanagementprozess (IEC 62304 Kapitel 8) und den Software-Wartungsprozess (IEC 62304 Kapitel 6) durchlaufen. Als Folge müssen Sie
- explizit freigeben, dass die Software geändert werden darf,
- Ursachen für diese Änderung erforschen (diese können z. B. im Prozess, der Anwendung von Methoden und Werkzeugen und nicht nur in einer fehlerhaften Code-Zeile liegen),
- Trends analysieren,
- die neuen/geänderten Software-Anforderungen dokumentieren, ebenso mögliche Änderungen an der Software-Architektur,
- je nach Sicherheitsklasse die Tests (Unit-Tests, Integrations- und Systemtests) als Regressionstests wiederholen (mehr zu weiter unten) und
- die geänderte Software freigeben.
Forderungen der FDA: Design Change
Die Forderungen der FDA sind vergleichbar den europäischen. Die FDA verlangt bei Design-Changes:
Each manufacturer shall establish and maintain procedures for the identification, documentation, validation or where appropriate verification, review, and approval of design changes before their implementation.
Besonders sollten Sie darauf achten, dass Sie die einzelnen Entwicklungsstände konsistent im Design History File DHF dokumentieren.
Software-Wartung: Praxistipps
Software-Wartung während der Entwicklung
Was tun, wenn während der Entwicklung ein Fehler auffällt beispielsweise
- beim Review eines Dokuments wie einer Software Requirements Specification oder der Software-Architektur?
- beim Code-Review?
- während des Programmierens?
- beim Software-Testing (z. B. Unit-Testing, Integrations- oder System-Testing)?
Streng genommen, müssen wir drei verschiedene Prozesse der IEC 62304 beachten:
- Problemlösungsprozess
- Konfigurationsmanagementprozess
- Wartungs- bzw. Entwicklungsprozess
Gehen wir auf diese drei Prozesse ein und diskutieren, wie wir das ohne Overhead hinbekommen:
ad 1. Problemlösungsprozess
Gesetzliche Forderung: Die wesentliche Forderung besteht darin, dass Sie das Problem dokumentieren und bewerten. Die Problemberichte müssen Sie im Rahmen der kontinuierlichen Messung und Verbesserung regelmäßig auswerten und Trends analysieren.
Praxistipp: Das können Sie mit einem einfachen „Ticketsystem“ realisieren. Das einfache Erfassen des Problemberichts sollte eine Sache von Minuten sein. Tragen Sie am besten gleich die Dokumente ein, die Sie ändern wollen.
Diese Problemlösungsprozess sowie die folgenden Prozesse müssen Sie aber nur durchlaufen, wenn ein bereits freigegebenes Dokument oder getestetes Artefakt nochmals geändert werden muss. Wenn der Entwickler beispielsweise beim Unit-Test feststellt, dass sein eben geschriebener Code nicht fehlerfrei ist, gibt es keine Notwendigkeit, einen Problembericht zu schreiben.
ad 2. Konfigurationsmanagementprozess
Gesetzliche Forderung: Das Ziel des Konfigurationsmanagements besteht v. a. in der Erlaubnis, das Dokument (das bezieht sich auch auf Code) zu ändern. Diese Freigabe muss die im Konfigurationsmanagementplan identifizierte Person erteilen.
Praxistipp: Sorgen Sie dafür, dass diese Person gut verfügbar ist. Nutzen Sie ggf. das gleiche „Ticket-System“, mit dem diese Person ihr Okay erteilt und dabei auch die Dokumente benennt, die geändert werden dürfen. Auch das sollte eine Sache von Minuten sein. Erteilen Sie die Freigabe nicht für einzelne Code-Dateien sondern für eine ganze Software-Einheit oder Software-Komponente.
ad 3. Software-Wartung
Jetzt durchlaufen die Änderungen den normalen Entwicklungsprozess. Sie legen fest, welche Teile Ihrer Software neu getestet werden müssen (s. u. Regressionstests).
Allgemeine Tipps
Meist scheuen sich Hersteller, während der Entwicklung Änderungen diesen drei Prozessen zu unterwerfen. Das liegt meist daran, dass die Freigabe der Änderungen und die Freigabe der geänderten Dokumente bzw. Software zu aufwendig ist. Dies ist aber ein Problem Ihrer Prozesse. Achten Sie darauf, dass
- Ihre Verfahrensanweisungen nur die Personen an den Prüfungen und Freigaben beteiligt, die
- wirklich davon betroffen sind und eine Ahnung davon haben. Manager zählen nicht notwendigerweise dazu.
- schnell verfügbar sind.
- Sie klare Kriterien für die Prüfungen und Freigaben haben, damit man genau weiß, was zu tun ist.
- Ihr Risikomanagement und Ihre Software-Architektur bekannt und dokumentiert sind und Sie darauf basierend den Umfang der Regressionsprüfung bestimmen können.
- Änderungen klar erkennbar sind (z. B. Überarbeiten-Modus in Word oder Diff im Versionsverwaltungssystem), um den Umfang für die Regressionsprüfungen zu minimieren.
Sie wollen Ihre Prozesse sehr effektiv und effizient gestalten. Das bedeutet, dass Sie auch experimentieren und Ihre entsprechenden Verfahrensanweisung regelmäßig reflektieren und überarbeiten. Für die Freigabe dieser Verfahrensanweisungen gilt übrigens das oben Gesagte.
Klären, ob es überhaupt eine Software-Wartung ist
Manche Hersteller hadern damit, ob eine Änderung beispielsweise an einer Sprachdatei oder Konfigurationsdatei überhaupt eine Software-Änderung darstellt, die dem Software-Wartungsprozess unterliegt. Die Antwort auf diese Frage hängt davon ab, ob die Änderungen dieser Dateien Teil der Zweckbestimmung (genauer gesagt des bestimmungsgemäßen Gebrauchs) sind.
Wenn beispielsweise eine Konfigurationsdatei dazu dienen soll, Netzwerk-Settings für eine Software-Installation vom Kunden oder Service-Techniker anzupassen, dann wäre das nicht als Software-Wartung zu verstehen. Vielmehr müsste bereits im Risikomanagement untersucht worden sein, was passiert, wenn jemand dabei ein Fehler unterläuft oder die Datei aus anderem Grund fehlerhaft ist.
Wenn hingegen die Konfigurationsdatei eine Datei ist, in die die Software-Entwickler Parameter ausgelagert haben, die aber nicht dazu bestimmt ist, von Dritten angepasst zu werden, wäre eine Änderung ein „Design Change“.
Neueinreichung der technischen Dokumentation bei einer Benannten Stelle
Wie groß die Änderungen maximal sein dürfen, damit man auf eine erneute Einreichung verzichten kann, unterscheidet sich von Benannter Stelle zu Benannter Stelle. Manche erwarten selbst bei kleinen Bugfixes ein Update der Akte, manche sagen eher „verschont uns!“.
Sie können mit folgender Faustregel arbeiten: Ein Bug-Fix (typischerweise dritte Stelle der Versionsnummer z. B. 2.12.4) bedarf keiner Meldung, es sei denn Sie müssen gemäß der Vigilanz-Anforderungen der MDR eskalieren. Funktionserweiterungen, neue Bildschirmmasken und Änderungen der Architektur (Design Change) bedingen eine neue Einreichung.
Stellen Sie sich immer die Frage, wenn Sie unsicher sind, ob eine Neueinreichung notwendig ist, ob die Änderungen eine wenn auch noch so kleine Änderung des Risikos bedeutet. Falls dem so ist, dann reichen Sie bitte neu ein.
Bei Medizinprodukten der Klasse I ist generell keine Einreichung der technischen Dokumentation vorgeschrieben — weder initial noch bei Änderungen des Produkts.
Risikomanagement
Im Risikomanagement müssen Sie darüber nachdenken, welche Folgen die Änderung nach sich ziehen kann und welche Risiken dadurch entstehen. Das sind
- Risiken durch die Änderung per se z. B. durch neue Funktionalitäten,
- Risiken, die sich durch neu eingeführte Bugs ergeben,
- Risiken durch die (erneute) Installation bzw. das Update der Software (beim Kunden) und
- Risiken durch mangelnde Gebrauchstauglichkeit, beispielsweise, weil die Nutzer die neuen Funktionen nicht kennen oder missverstehen.
Gleich ob Sie die Zweckbestimmung, die Software Requirements Specification (einschließlich Spezifikation der Benutzerschnittstelle), die Software-Architektur oder den Code ändern: Die Risikomanagementakte wollen Sie entsprechend aktualisieren.
Abhängig von den neuen Risiken, die sich durch die Software-Wartung ergeben, werden Sie Maßnahmen ergreifen. Zu diesen Maßnahmen kann auch eine erneute Schulung der Benutzer zählen. Dazu sind auch die Betreiber gemäß MPBetreibV verpflichtet.
Software-Tests und Regressionstests
Im Rahmen des Software-Tests müssen Sie nachweisen, dass
- die neue bzw. geänderte Funktionalität (d. h. die neuen oder bisher nicht umgesettzen Software-Anforderungen) wirklich wie spezifiziert implementiert wurde und
- die davon nicht betroffenen Software-Anforderungen weiter wie spezifiziert erfüllt sind.
Im einfachsten Fall testen Sie bei einer Änderung die Software nochmals vollständig. Das wird i.d.R. nur dann möglich sein, wenn die Software-Tests vollständig automatisiert sind. Falls das nicht möglich ist, dann testen Sie „nur“ die neuen Funktionalitäten und die Funktionalitäten, die von der Änderungen betroffen sein können. Welche das sind, können Sie nur in der Software-Architektur erkennen bzw. damit begründen.
Erneute Überprüfung der Gebrauchstauglichkeit bei der Software-Wartung
Die IEC 62366 verlangt eine Validierung der Gebrauchstauglichkeit. Bei einer Software-Wartung würden Sie die Benutzungsszenarien (erneut) testen, im Rahmen derer geänderte Benutzer-Produktschnittstellen (z. B. eine GUI) durchlaufen werden.
Denken Sie daran, dass Sie diese Gebrauchstauglichkeitsvalidierung mit repräsentativen Nutzern (Plural!) in einer repräsentativen Nutzungsumgebung durchführen.
Prozesse gesetzeskonform gestalten
Wünschen Sie Unterstützung dabei,
- Ihre Prozesse, z. B. den Software-Wartungsprozess neu und gesetzeskonform zu gestalten,
- Werkzeuge auszuwählen und anzupassen, um diese Prozesse effektiv und effizient zu unterstützen oder
- Ihr QM-Handbuch, Ihre Prozessbeschreibungen und Ihre Dokumente (z. B. auch das Design History File) zu prüfen, um Problem im Audit zu vermeiden?
Dann nehmen Sie mit uns Kontakt auf! Wir sind darauf spezialisiert und können oft innerhalb weniger Stunden helfen.
Änderungshistorie
- 2024-01-19: Verweise auf die MDR ergänzt
- 2015-09-11: Initiale Erstellung des Artikels


Hallo Herr Pr. Dr. Johner,
vielen Dank für den informativen Artikel. Diesbezüglich hätte ich noch einige Fragen:
Meinem Verständnis nach verstehe ich die Software-Wartung als Update. Hier wird von Medizinprodukten gesprochen, doch wie sieht es mit IVDs aus? Und wie verhält es sich mit Updates einer embedded SW in einem IVD? Die Klassifizierung würde unter andere IVD fallen (IVDD), bei dem keine Benannte Stelle involviert ist. Demnach muss keine Meldung nach einem Update erfolgen, sondern nur die Aktualisierung aller relevanten technischen Dokumente. Verstehe ich das so richtig?
Viele Grüße
Lisa N.
Hallo Lisa N.,
genau, unter Software-Wartung versteht man die Aktivitäten nach dem ersten Freigeben des Software-Systems, z.B. Updates/Hot-Fixes. Aus Sicht der Regularien ist es egal, ob es „nur“ ein Medizinprodukt oder auch ein IVD und ob es eine Standalone oder Embedded Software ist: Änderungen sollen kontrolliert erfolgen, Risiken und Seiteneffekte in Bezug auf die Änderung analysiert werden und je nach Ergebnis der Analyse andere Abteilungen, Risikomanager, PRRC, Benannte Stelle und/oder Behörden informiert werden. Wenn in ihrem Konformitätsbewertungsverfahren keine Benannte Stelle involviert ist, haben Sie natürlich auch keine Pflichten diesen gegenüber. Die Technische Dokumentation gemäß Anhang II/III IVDR bzw. Anhang III IVDD müssen Sie aber immer auf Stand halten.
Viele Grüße
Daniel Reinsch
Dear Mr. Reinsch,
As mentioned in the comment above, „if no notified body is involved in your conformity assessment procedure, you have no obligations towards them“ as regards to SW maintenance, even if these are updates. However, given the current IVDR and art.110 (3) what happen to legacy devices, which are not subjected to NB-conformance assessment originally, but should do upon IVDR transition? And if the SW maintenance-fixes/updates involve changes that could affect significantly the SW design? Is still unnecessary to involve a NB or a CA?
Thank you and with best regards,
David S.
Dear David,
when the software changes are significant, then you have to be fully compliant with the new regulation. That means that you need to involve a notified body (as required by the conformity assessment procedure of the IVDR corresponding to your IVDR risk class). Minor changes, it-security patches, and bug fixes can still be done during the transitional period without completely fulfilling all requirements of the new regulation. These non-significant changes can be done without involving a notified body if your conformity assessment procedure under IVDD didn’t involve one.