
Definition des Begriffs UOUP (gemäß IEC 62366)
Die IEC 62366-1:2015 (die Norm zur „Anwendung der Gebrauchstauglichkeit auf Medizinprodukte“) führt den Begriff UOUP ein und definiert ihn wie folgt:
| Inhaltsübersicht |
|---|
| Ziele » |
| Erleichterungen » |
| UOUP und SOUP » |
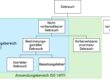
Was die IEC 62366 mit dem Konzept der UOUP erreichen will
Viele Medizinprodukte-Hersteller kaufen Komponenten wie z.B. eine Computer-Maus zu, die offensichtlich keinen „Usability Engineering Process“ gemäß IEC 62366 durchlaufen hat. Oder sie haben Altprodukte seit Jahrzehnten ohne Probleme im Markt oder möchten für diese eine kleinere Änderung an der UI machen.

Nun müssten die Hersteller eigentlich für diese Benutzerschnittstellen den kompletten Prozess nachträglich durchlaufen. Genau da möchte ihnen die neue IEC 62366 entgegenkommen: Sie dürfen einen verkürzten Usability Engineering Prozess durchlaufen, bei dem v.a. die Risiken bewertet werden, die mit der Gebrauchstauglichkeit dieser Komponente einhergehen. Dabei muss der Hersteller auch auf Informationen zurückgreifen, die aus dem Markt bereits vorhanden sind.
Ganz genau kann dieser verkürzte Prozess für drei Szenarien angwendet werden:
- Auf (Legacy) Medizinprodukte, die bereits auf dem Markt sind und die nicht unter Verwendung des Prozesses gemäß IEC 62366:2006 (sogenannte Vorgänger-Medizinprodukte) entwickelt wurden
- Auf (Legacy) Medizinprodukte, die nur kleine Änderungen aufweisen und die ursprünglich nicht unter Verwendung von IEC 62366:2006 entwickelt wurden
- Auf Medizinprodukte, die eine handelsübliche Komponente enthalten, die ihrerseits nicht unter Verwendung gemäß IEC 62366:2006 entwickelt wurde. Ein Beispiel war die o.g. Computer-Maus.
Welche Erleichterungen die Norm erlaubt
Erleichterungen durch die IEC 62366:2006
Die Erleichterungen bestehen im Wesentlichen darin, dass einige der Schritte des gebrauchstauglichkeitsorientierten Entwicklungsprozesses (Usability Engineering Process), wie von der IEC 62366:2006 vorgeschrieben, weggelassen werden können. Das betrifft insbesondere
- die Usability Spezifikation
- die Usability Verifizierung
- den Usability Validierungsplan
- die Usability Validierung selbst.
Dafür müssen die Rückmeldungen in jeder Form daraufhin ausgewertet werden, ob sie Informationen zu Risiken durch mangelnde Gebrauchstauglichkeit enthalten. Als Informationsquellen eigenen sich:
- Berichte vom Service
- Kundenbeschwerden
- Meldungen bei und von Behörden
- Anrufe bei Hotline
- Beobachtungen
Nicht auslassen dürfen die Hersteller aber die sogenannte „Application Specification“ also die Zweckbestimmung sowie die Feststellung der Hauptbedienfunktionen.
Erleichterungen durch die IEC 62366-1:2015
Bei den Erleichterungen gemäß IEC 62366-1:2015 sieht es vergleichbar aus: Hier darf auf die formative und summative Bewertung, sogar auf die Beschreibung der „Use Scenarios“ verzichtet werden, aber nicht auf die Festlegung der „Use Specification“.
UOUP: Der Zeitpunkt entscheidet
Eine Ergänzung von Wolfgang Schneider, Usability Experte am Johner Institut und Mitglied der Arbeitsgruppe zur IEC 62366-1Die Norm IEC 62366-1:2015 spricht im normativen Anhang C sehr explizit vom Publikationsdatum der Norm, das herangezogen werden sollte, um zu beurteilen, ob ein UI unter UOUP subsumiert werden kann oder nicht – also das Jahr 2015.
Normativer Text
Der entscheidenden Sätze lauten:
C.1 General
This annex was created in recognition of the fact that many MANUFACTURERS will be interested in applying the tools defined in this standard to USER INTERFACES or parts of USER INTERFACES that have already been commercialized prior to the publication of this edition of this standard.
[…]
However, if any modifications are made to the USER INTERFACE or its parts, only the unchanged parts of the USER INTERFACE remain UOUP and the changed parts of the USER INTERFACE are subject to 5.1 to 5.8.
Schlussfolgerung
Es betrifft also tatsächlich das Jahr 2015, wenn der Hersteller die Version 2015 der Norm verwenden möchte. Zudem ist der Anhang C definitiv „normative“ und nicht „informative“, im Gegensatz z.B. zum Anhang B. Somit gilt das Publikationsdatum. Im neuen AMD-Draft ist auch vorgesehen, das Datum 2015 im Anhang C zu ergänzen, wo es noch nicht erwähnt wurde.
Wenn der Hersteller noch die harmonisierte Vorgängerversion nehmen möchte, gilt Anhang K und das alte Jahr 2007.
Daher dürfte es für die Hersteller interessanter sein, nicht das „alte“ Erscheinungsdatum und die harmonisierte Norm zu verwenden, sondern das neue mit 2015 und den Anhang C. Da dürften dann mehr Produkte darunter fallen.
Da die Norm verlangt, dass Hersteller die UI-Teile, die nach der Veröffentlichung entwickelt oder verändert wurden, nach dem Prozess der 62366-1 entwickeln soll, sind sie de facto gezwungen, das Publikationsdatum als Kriterium heranzuziehen.
Hinweis: Der Prozess im Anhang C kann als optional eingestuft werden, denn im normativen Teil wird nur von „may be evaluated according to Annex C” gesprochen. Das heißt, es gibt keine Verpflichtung („shall“) den Prozess im Anhang C so zu nutzen. Ein Widerspruch der Norm.
Fazit
Die Empfehlung lautet, sich auf das Publikationsdatum der aktuellen 62366-1 zu beziehen, so wie es im Anhang C steht, wobei der Anhang C durch das Korrigendum von 2016 und das neue AMD nochmals bestätigt wird.
Weitere Informationen zu UOUP
UOUP (User Interface of Unknown Provenance) und SOUP (Software of Unknown Provenance)
Für diese „bewährten“ „User Interfaces of unknown provenance“ hat man den Ansatz aus der IEC 62304 übernommen, die sogenannte „Software of Unknown Provenance“ SOUP kennt. Sowohl für die Software als auch die Benutzerschnittstellen unbekannter Herkunft wird auf den sonst vorgeschriebenen Umfang an Dokumentation verzichtet, wenn sich diese bewährt haben und der Hersteller die Risiken als akzeptabel bewertet.
Konsequenzen
Das Konzept der UOUP ist nicht auf „User Interfaces“ von Dritten beschränkt, sondern lässt sich auch auf eigene Produkte bzw. Benutzerschnittstellen davon anwenden, die in Verkehr gebracht wurden, bevor es die IEC 62366 gab und die sich im Markt bewährt haben. Sobald ein Hersteller aber Änderungen an diesen User Interfaces vornimmt, muss er wieder den von der IEC 62366 vorgeschriebenen „Usability Engineering Process“ in Gänze durchlaufen. D.h. über Dokumentationssünden in der Vergangenheit sieht man etwas hinweg, wenn dadurch keine Risiken für Patienten einhergehen. Aber ein Freibrief für die weitere Entwicklung von „Medical Devices“ ist das definitiv nicht.
UOUP und FDA
Bereits bevor die IEC 62366-1:2015 erschien, gab es ein Amendment in Form eines Annex K, das genau dieses Konzept der UOUP nachträgt, das übrigens von der FDA befürwortet wird. Das ist gut, denn sonst wäre dieses „Entgegenkommen“ für alle Medizinprodukte-Hersteller, die auch in den US-amerikanischen Markt streben, wertlos.

Ist unter dem Abschnitt
„Was die IEC 62366 mit dem Konzept der UOUP erreichen will“, bei Punkt 1 – 3, wirklich die IEC 62366:2007 oder doch die IEC 62366-1:2015 gemeint ?
Schönen Gruß
Das gilt bereits ab dem Ammendment zur „alten“ IEC 62366. Die neue Version behält das UOUP Konzept bei.
Zu dem Abschnitt: „Nicht auslassen dürfen die Hersteller aber die sogenannte „Application Specification“ also die Zweckbestimmung sowie die Feststellung der Hauptbedienfunktionen.“
Ist es sicher, dass die Hauptbedienfunktionen, tatsächlich zu UOUP gehören? In der 62366-1 kann ich aus der UOUP Sektion die Notwendigkeits zur Hauptbedienfunktionsbestimmung nicht herleiten.
Sie haben völlig Recht, aus der 62366-1 folgt das nicht.
Der Beitrag stammt noch aus dem Jahr 2015 und bezieht sich auf die alte Version bzw. den Anhang K.
Ich werde den Beitrag aktualisieren. Danke für Ihren wertvollen Hinweis, Herr Fiege!
Um die Frage von Herrn Schmidberger nochmals aufzugreifen:
Woher genau kommt denn die Einschränkung „und die nicht unter Verwendung des Prozesses gemäß IEC 62366:2007 (sogenannte Vorgänger-Medizinprodukte) entwickelt wurden“ aus den Aufzählungspunkten 1.-3. ?
In der IEC62366-1:2015 steht im Annex C, dass der in diesem Annex beschriebene Prozess für User Interfaces anwendbar ist, für die keine Doku nach IEC62366-1 vorhanden ist. Die IEC62366:2007 wird nicht erwähnt.
Und den Annex K gibt es, soweit ich das nachvollziehen konnte, auch erst seit dem IEC62366 Amendment A1 von 2014.
Meine eigentlich Frage wäre:
Wenn ich nun eine Doku nach IEC62366:2007 vorliegen habe, für ein Gerät, dass 2010 entwickelt wurde und nicht geändert wird, kann ich mich dann auf Annex C berufen?
Vielen Dank im Voraus für jegliche Hilfe bei dem Thema!
Lieber Herr Kochanski,
Danke für die gute Frage!
Ich denke, die Beschränkung auf die IEC 62366-1 ist ein redaktioneller Fehler. Ich frage bei der nächsten Sitzung des Normenkomitees nach.
Wenn Sie eine Dokumentation haben, die den Forderungen der IEC 62366:2007 entspricht, gleich ob das Produkt vor oder nach 2010 in Verkehr gebracht wurde, bedarf man des Annex C nicht. Schließlich ist das immer noch die aktuell harmonisierte Norm.
Wenn Sie oder Ihre benannte Stelle die IEC 62366-1 als den Stand der Technik definieren, was sicher eine gute Idee ist, dann bedürfen Sie des Anhangs C auch nicht. Schließlich wurde das Produkt legal in Verkehr gebracht. Im Rahmen der Marktüberwachung müssen Sie sowieso auch Risiken durch Gebrauchstauglichkeit kontinuierlich neu bewerten und ggf. Maßnahmen ergreifen.
Der Annex C wird für Sie spannend, wenn Sie ein neues Produkt mit alter UI in den Markt bringen.
Hilft das, falls nicht, gerne einfach nachhaken, gerne auch direkt per E-Mail.
Christian Johner
Sehr geehrter Herr Prof. Johner,
ich möchte noch einmal auf Ihre letzten Eintrag vom 28.02.2017 zurückkommen.
Insbesondere geht es um:
„(…) Ich denke, die Beschränkung auf die IEC 62366-1 ist ein redaktioneller Fehler. Ich frage bei der nächsten Sitzung des Normenkomitees nach. (…)“
Konnten Sie zwischenzeitlich schon klären, ob es sich um einen redaktionellen Fehler handelt oder darf ich nun die UOUP-Anforderungen auch in der Nachdokumentation von Produkten benutzen, die ich zwischen 2007 und 2015 erstmals in den Verkehr gebracht habe.
Über Ihre Rückmeldung würde ich mich sehr freuen.
MFG
Werner Knopp
Sehr geehrter Herr Knopp,
es ist mir super peinlich, aber ich hatte Ihre Anfrage von vor zwei Jahren vergessen gehabt. Ich bin mir zwar ziemlich sicher, dass das UOUP-Konzept für legal(!) vermarktete Geräte gedacht ist und nicht zum „Heilen“ von Gesetzesverstößen. Aber ich frage nach, wie haben im Februar die nächste Sitzung.
Mit bestem Denk für Ihr Erinnern, mit der Bitte um Nachsicht für mein Vergessen und mit herzlichen Grüßen
Christian Johner
Sehr geehrter Herr Johner,
Sie schreiben „Bereits bevor die IEC 62366-1:2015 erschien, gab es ein Amendment in Form eines Annex K, das genau dieses Konzept der UOUP nachträgt, das übrigens von der FDA befürwortet wird. “ Worauf genau beziehen Sie sich, wenn Sie sagen, dass die FDA das Vorgehen befürwortet? Gab es hierzu bereits eine schriftliche Äußerung der FDA?
Vielen Dank und beste Grüße,
Franziska Herrmann
Die FDA hat im Human Factors Engineering Guidance ebenfalls argumentiert, dass die Post-Market Erfahrungen mit einbezogen werden dürfen/sollen.
Sehr geehrter Herr Johner,
bei chirurgischen Instrumente, deren Entwicklung viel weiter als 2006 zurückliegt, würde es demnach bedeuten, dass diese als UOUP-Medizinprodukte betrachtet werden können? Gilt dies auch, wenn der „Hersteller“ die chirurgischen Instrumente nicht eigens entwickelt hat und diese vom originären Hersteller bezieht und unter eigenem Namen in Verkehr bringt? Wie verhält es sich, wenn der originäre Hersteller die chirurgischen Instrumente auf Grundlage der einschlägigen Normen herstellt, also den Stand der Technik nutzt (keine eigene Entwicklung)?
Mit freundlichen Grüßen
Philipp Kienel
Sehr geehrter Herr Kienel,
sehr gerne antworte ich an Herrn Johners Stelle.
Wenn ich Sie richtig verstehe, beschreiben Sie ein OEM/PLM-Konstrukt. Auch in diesem Fall würde das UI als UOUP gelten, sofern es nach Erscheinen der IEC 62366-1:2015 nicht verändert wurde. Der UOUP-Prozess nach Anhang C der IEC 62366-1 sollte korrekt befolgt und dokumentiert sein. Wichtig für Sie als PLM ist, das Sie vollen Zugriff auf die entsprechende Technische Dokumentation des „originären Herstellers“ haben (siehe OEM (Original Equipment Manufacturer) von Medizinprodukten).
Vielen Dank für Ihre Frage und herzliche Grüße
Nils Becker
Sehr geehrter Herr Prof. Johner,
wir haben bei uns im Unternehmen eine Rauchgasabsaugung im Portfolio.
Streng genommen sind es zwei Produkte/zwei Produktakten: marVac und smartVac
Die marVac wurde im Januar 2011 fertig entwickelt.
Die smartVac im Dezember 2016.
Die beiden Geräte sind exakt gleich: Gleiche Leistungsdaten mit gleichem User Interface. Somit ergeben sich aus Usability Sicht keinerlei Unterschiede.
Der einzige Unterschied sind die äußeren Gehäuseteile, welche minimal andere Abmessungen haben. Unterschied max. 1 cm. Das Design der smartVac sieht etwas moderner aus.
Können wir trotzdem nach UOUP vorgehen? Die marVac würde zeitlich ja passen, die smartVac leider nicht.
Die für die Usability relevanten Punkte sind wie gesagt identisch.
Vorab vielen Dank für Ihre wertvolle Rückmeldung hierzu.
Freundliche Grüße
Ralf Kohler
Sehr geehrter Herr Kohler,
vielen Dank für Ihre spannende Frage.
Anhang C der IEC 62366-1 ermöglicht es Herstellern einen schlankeren Prozess für User Interfaces, die vor 2015 bereits Inverkehr gebracht worden sind, anzuwenden. Da das Produkt marVac mit einem nach Ihrer Aussage identischen User Interface zu smartVac bereits vor 2015 entwickelt und in den Verkehr gebracht wurde, kann der UOUP-Prozess auch für das User Interface für smart Vac angewendet werden. Der entscheidende Punkt ist der Zeitpunkt der Inverkehrbringung des User Interfaces und nicht des gesamten Produktes. Wurden oder werden allerdings nach 2015 signifikante Änderungen am User Interface vorgenommen oder falls sich die Zweckbestimmung und insbesondere die Benutzergruppen von den beiden Produkten unterscheiden, müssen diese Modifikationen den Usability-Prozess nach Kapitel 5.1 – 5.9 der Norm durchlaufen. Die Norm listet in Anhang C.1 ein paar anschauliche Beispiele dafür.
Ich hoffe meine Antwort hilft Ihnen weiter. Ansonsten haken Sie gerne nach.
Herzliche Grüße
Nils Becker
Sehr geehrter Herr Dr. Becker,
vielen Dank für Ihre hilfreiche Antwort, das hilft uns sehr weiter.
In der Norm haben wir folgenden Satz gefunden: „Die Begleitdokumentation wird als Teil des Medizinprodukts und seines User Interface betrachtet.“
Wie verhält es sich, wenn über die Jahre hinweg, teilweise auch nach 2015, die Gebrauchsanweisung revisioniert wurde? Es wurden kleinere Anpassungen vorgenommen, neue Templates verwendet, etc.
Die Änderungen dürften keinen Einfluss auf die Usability bzw. die Handhabung des eigentlichen User Interface gehabt haben.
Vielen Dank.
Herzliche Grüße
Ralf Kohler
Sehr geehrter Herr Kohler,
es freut mich sehr, dass ich Ihnen weiterhelfen konnte.
Die IFU zählt, wie die IEC 62366-1 schreibt, ebenfalls zum UI und die Erstellung dieser bzw. Änderungen müssen somit ebenfalls den Usability-Engineering-Prozess durchlaufen. Änderungen an der IFU nach Inverkehrbringung sollten einer Risikoanalyse unterzogen werden. Könnten die Änderung einen Einfluss auf die sichere Gebrauchstauglichkeit des Produkts haben? Wird das Design der IFU so stark verändert, dass nicht mehr zweifelsfrei gesagt werden kann, ob wichtige Information entdeckbar, auffindbar und verständlich sind? Werden vielleicht sogar neue Risikokontrollmaßnahmen als Informationen zur Sicherheit in der IFU implementiert? Falls ja, müssten Sie im Zweifel die Änderungen mit den vorgesehenen Benutzergruppen summativ evaluieren. Falls die Änderungen nicht risikorelavant sind, müssten Sie dies nur in Ihrer technischen Dokumentation entsprechend darlegen.
Bei weiteren Fragen schreiben Sie mir gerne.
Herzliche Grüße
Nils Becker