
ISO 14971:2012 (Anhang ZA): Die wichtigsten Änderungen der Norm
Quasi über Nacht, nämlich vom 31.08.2012 zum 01.09.2012 wurde die ISO 14971:2012 ohne Übergangsfrist als harmonisierte Norm für das Risikomanagement für Medizinprodukte veröffentlicht. Dieser Artikel stellt Ihnen diese Änderungen vor.
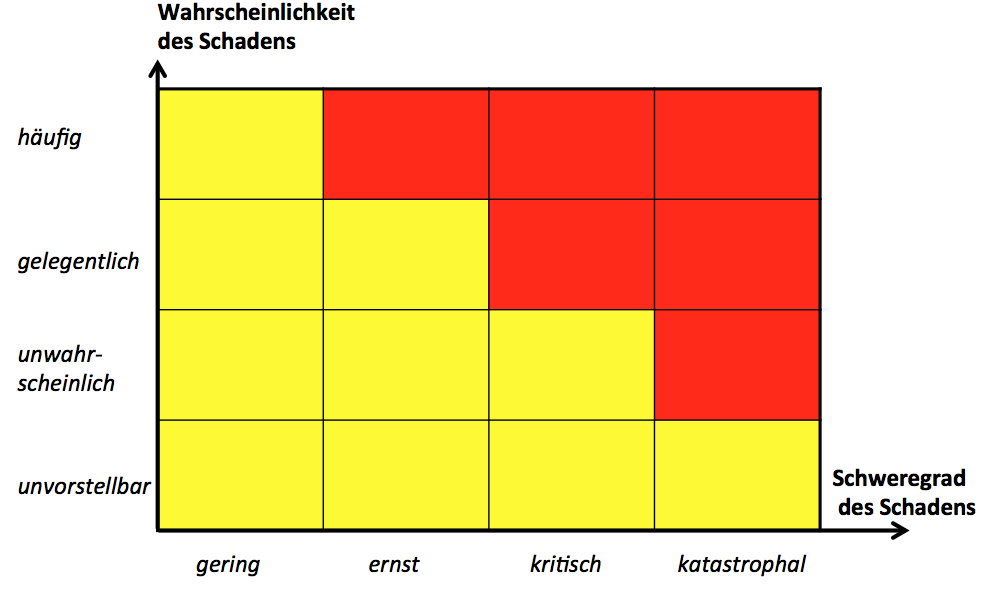
Mit den Änderungen durch die ISO 14971:2012 gibt es keine prinzipiell akzeptablen Risiken mehr.
Eine der wesentlichen Neuerungen der Norm ISO 14971 im Jahr 2012 (ISO 14971:2012) besteht in der Anforderung, dass generell alle Risiken so weit wie möglich zu minimieren sind, es also keine per se akzeptablen Risiken gibt. Die Klarstellungen und Ergänzungen im informativen(!) Anhang ZA empfinde ich teilweise als Neudeutung des bisher Geschriebenen. Gehen wir die einzelnen Teilkapitel dieses Anhangs durch:
- Kapitel: Während es bisher „generell“ vernachlässigbare Risiken gab, sind diese jetzt auf einmal verschwunden. Jedes einzelne(!) Risiko ist zu bewerten. Das bedeutet, dass es keine Risikoakzeptanzmatrix aus akzeptablen, ALARP und nicht akzeptablen Risiken (also „grün, gelb und rot“) mehr geben kann, sondern nur noch gelbe und rote Risiken gibt. Das ist wirklich etwas Neues.
- Kapitel: Es existiert insbesondere keine Schwelle mehr, unterhalb derer ein Hersteller nichts tun müsste, um Risiken zu minimieren. Es existiert jetzt nur noch eine Schwelle, unterhalb derer die bereits reduzierten Risiken akzeptiert werden können. Das ist ein Unterschied!
- Kapitel: Weiter verlangt die Norm, dass Risiken nicht mehr nur „as low as reasonable practical“ reduziert werden dürfen, sondern „as low as reasonable possible“ reduziert werden müssen. Der Unterschied besteht in ökonomischen Überlegungen, die in der zweiten Version keine Rolle mehr spielen dürfen.
- Kapitel: Im alten Verständnis der ISO 14971 war die Gesamt-Risiko-Nutzenbewertung eigentlich nur dann erforderlich, wenn nicht alle Risiken im grünen Bereich lagen. Diese Gesamtbewertung, die alle Risiken umfasst, ist spätestens jetzt mit der ISO 14971:2012 immer Pflicht.
- Kapitel: Die „alte 14971“ hatte man so verstehen können, dass man zwischen den Maßnahmen „inhärente Sicherheit“, „Schutzmaßnahme“ und „Information“ wählen könne, so lange man in dieser Reihenfolge versucht, die Maßnahmen umzusetzen. Nun wird klargestellt, dass alle Maßnahmen – so möglich – kumulativ anzuwenden sind.
Weshalb eine inhärent sichere Maßnahme aber weiterer Maßnahmen bedarf, erschließt sich mir nicht. Darauf zu verzichten, zuerst inhärente Maßnahmen anzustreben, war hingegen schon immer im Widerspruch zur Norm. - Kapitel: Dieses Kapitel schließt direkt an das eben Gesagte an und wiederholt, dass die MDD von „inherently safe design and construction“ spricht. Das eigentliche Problem, nämlich die fehlende Definition der inhärenten Sicherheit, wird aber weiter nicht behoben.
- Kapitel: Die Informationen an den Nutzer dürfen nicht länger als risikominimierende Maßnahmen verstanden werden. Das halte ich erneut für missverständlich. Natürlich verschwindet ein Risiko nicht dadurch, dass man einen Nutzer generell über das Risiko informiert. Wenn jedoch, um ein Beispiel zu nennen, ein Medizinprodukt bei der Eingabe eines Medikaments den Benutzer fragt, „Sind Sie sicher, dass Sie Herrn XY ein Penicillin trotz dessen Penicillin-Allergie verschreiben wollen?“, dann ist das durchaus eine Maßnahme, die geeignet ist, das Risiko durch das Produkt zu reduzieren, hier ein falsches Medikament damit zu verschreiben. Die Reduktion des Risikos ergibt sich aus der Reduktion der Wahrscheinlichkeit.
Der Auditgarant zeigt Ihnen über 15 Videotrainings Schritt für Schritt, wie Sie eine schlanke ISO 14971:2012 konforme Risikomanagementakte erstellen.
Wie es zur ISO 14971:2012 kam
Forderung der Medizinprodukte-Richtlinie
Harmonisierte Normen dienen dazu, Herstellern konkrete Hinweise zu geben, wie sie Forderungen der Richtlinien einhalten. Erfüllen die Hersteller die Normen, vermutet man, dass sie auch die Forderungen der Richtlinien erfüllen.
Das wichtigste Anliegen – vielleicht sogar das einzig relevante – besteht darin, dass Medizinprodukte über ein akzeptables Risiko-/Nutzenverhältnis verfügen müssen. D.h. dass die Produkte den Patienten wirklich helfen und gleichzeitig die Nebenwirkungen so weit wie möglich reduziert werden müssen.
Die Probleme mit der (alten) ISO 14971
Der ALARP-Bereich im Risikomanagement ist definiert als der Bereich, bei dem Risiken „as low as reasonable practical“ (ALARP) reduziert werden müssen.Doch dieses „so weit wie möglich“ versteht die ISO 14971 ein wenig anders als die MDD – zumindest formuliert sie nicht ganz klar. Jedes Risiko gilt es „so weit wie möglich“ zu minimieren und nicht einige nur „as low as reasonable practical“. Was heißt schon „practical“? So weit, bis man keine Lust mehr hatte? So weit, bis man das Gerät neu designen müsste? So weit, bis keine Zeit und kein Geld mehr da sind?
Diese Praktikabilität hat in der Praxis viel mit ökonomischen Überlegungen zu tun. Und die haben im Risikomanagement nichts verloren. Eine Konsequenz könnte darin bestehen, diesen ALARP-Bereich einfach abzuschaffen. Eine andere darin, für jedes ALARP-Risiko zu begründen, weshalb eine Reduzierung nicht sinnvoll möglich ist. Also eher ein „as low as reasonable possible„. So handhabe ich das bei meinem Kunden schon die ganze Zeit, für die Klarheit bin ich aber dankbar.
Änderung der ISO 14971:2012 Welche Alternativen zur Verfügung standen
Doch was macht man, wenn sich herausstellt, dass eine Norm nicht mehr geeignet ist, diese Vermutung aufrechtzuerhalten? Beispielsweise, weil sie die Forderungen der Richtlinien unvollständig oder gar falsch adressiert? Man hat mehrere Möglichkeiten:
- Man deharmonisiert die Norm.
- Man ändert die Norm.
- Man interpretiert das bisher Geschriebene neu.
Für kurze Zeit wurde offensichtlich diskutiert, die ISO 14971 zu deharmonisieren(!). Nun hat man sich für die dritte Variante entschieden und einen Anhang zu ergänzen. Den Autoren der ISO 14971 gehen die Buchstaben für die Anhänge aus – wir sind jetzt bei ZA angekommen. An Ideen mangelt es Ihnen jedenfalls nicht. Denn das, was die 2012er Ausgabe von ihren Vorgängern unterscheidet, hat es in sich.
Sollten Sie Fragen zur praktischen Umsetzung der ISO 14971:2012 und den Neuerungen haben, dann nutzen Sie die kostenlose Auditsprechstunde.

Die Änhange mit dem Buchstaben ‚Z‘ kennzeichnen Anhänge, die speziell in der europäischen Version der jeweiligen Norm exisitieren. Dies ist zum Beispiel auch in der EN 60601-1 der Fall, während die IEC 60601-1 keine Z Anhänge besitzt!
Hallo Herr Dr. Johner,
mit Interesse habe ich Ihre Ausführungen zur ISO 14971 und zur Anwendung oder nicht Anwendung von Riskoakzeptanzkriterien gelesen. Ich kann aber bei einem Punkt nicht mitgehen. Sie schreiben: „Es existiert jetzt nur noch eine Schwelle, unterhalb derer die bereits reduzierten Risiken akzeptiert werden können. Das ist ein Unterschied!“
gemäß des Anhang ZA geht selbst das nicht: „Demzufolge darf der Hersteller vor Anwendung von Richtlinie 93/42/EWG, Anhang I, Abschnitte 1 und 2, keine Risiko-Akzeptanzkriterien anwenden. “
Für mich bedeutet das, ich kann Risikobewertungszahlen festlegen und eventuell farblich unterscheiden. Dies darf aber nur der Übersicht dienen. Eine Konsequenz darf ich nicht ableiten.
Was sagen Sie dazu?
Viele Grüße
Lutz Babilon
Sehr geehrter Herr Babilon,
ich stimme Ihren Aussagen zu, sehe aber den Widerspruch noch nicht ganz. Ich schreibe, wie Sie zitieren, dass Risiken unterhalb einer Schwelle akzeptiert werden können. Es geht um die Akzeptanz von Risiken nach den Maßnahmen. Man muss sicher Risiken akzeptieren, sonst kommt kein Produkt in den Markt. Und man wird sich überlegen, welche Risiken welcher Höhe das sind. Das meinte ich mit Schwelle.
Die Risikoprioritätszahl halte ich generell für ungeeignet (siehe https://www.johner-institut.de/blog/iso-14971-risikomanagement/risikoprioritaetszahl-rpz/). Insbesondere ist das Problem bei der RPZ, dass Sie „abstrakte“ Akzeptanzkriterien definiert, die nicht direkt den Nutzen und Schaden vergleichen. Und das kann nur pro Schweregradklasse erfolgen, nicht pauschal mit einem Grenzwert.
Falls ich Ihre Punkte noch nicht ausreichend adressiert haben sollte, dann schreiben Sie mir einfach (mail@johner-institut.de).
Viele Grüße
Christian Johner
Zum Thema „Warnungen und Hinweise, die eine Anleitung zur Vermeidung von Schäden sein sollen“:
Die Gebrauchsanweisung ist – genauso wie das Medizinprodukt selbst – ein Entwicklungsergebnis (Design Output). Dort ist verbindlich dokumentiert, was der Anwender tun muss respektive nicht tun darf. Selbstverständlich können Anweisungen risikomindernd sein, siehe Anhang J.2 der EN ISO 14971, „…was getan oder vermieden werden sollte, um Schaden zu verhindern.“
Wie sieht es aber bei der (Hintergrund-)Information über die Restrisiken aus? Gemäss Anhang J.3 der EN ISO 14971 sind Informationen über Restrisiken notwendig, „…um den Anwender über den sicheren und wirkungsvollen Gebrauch des Produkts zu informieren, zu motivieren und zu befähigen.“ Das heisst, auch solche Informationen können risikomindernd sein.
Formal ist es wichtig, dass der Anhang J von den Z-Anhängen nicht in Frage gestellt oder gar ausser Kraft gesetzt wurde. Betriebsintern fordert ja die EN ISO 13485 genau dasselbe: Gemäss Kapitel 6.2.2 d) ist das Hintergrundwissen der Mitarbeiter wichtig und wirksam um die Qualitätsziele zu erreichen; und unter den Qualitätzielen können durchaus auch Risikominderungen sein.
Diese Standpunkte haben wir gegenüber mehreren Benannten Stellen erfolgreich verteidigen können.
Besten Dank für die wichtigen Gedanken, Herr Angst!
Ihre Einschätzung bezüglich des Anhangs J teile ich.
Es gibt zwischen den benannten Stellen und den Behörden (konkret einer konkreten Person aus Brüssel) einen Dissens darüber, ob Hinweise als risikominimierende Maßnahmen dienen dürfen oder nicht.
Darüber, dass es dieser Hinweise bedarf, besteht Einigkeit.
Gilt das ALAP-Prinzip (As Low As Possible) gemäß Annex ZA denn auch nach Veröffentlichung der neuen Richtlinie 93/42/EWG? Ich sehe in meinem neuen Unternehmen noch die alte Umsetzung der ISO 14971:2003, welches ein erfolgreiches QMS zertifiziert hat.
Danke
Viele Grüße
Sarah
Mit der ISO 14971:2003 kann die Konformität mit den Anforderungen der 93/42/EWG nicht(!) mehr vermutet werden. Die Zertifizierung eines QMS ist zumindest tw. davon unabhängig, da es keine Zertifizierung nach ISO 14971 ist. Allerdings hätte beim „Tech File Review“ dieses Gap auffallen können / müssen.
Hallo Herr Johner, zwischenzeitlich ist auch die ISO/DIS 14971 erschienen:
https://www.beuth.de/de/norm-entwurf/din-en-iso-14971/290649384
Dies ist m.E. so interessant, da die ISO 14155 nur im Zusammenhang mit der ISO 14971 gültig ist und beide Drafts auch schon einen Z-Anhang mit Bezug zur MDR aufweisen. Wer seine Prozesse in Bezug auf die MDR gerade überarbeitet, sollte dies gleich mit aufnehmen! „Rund“ wird dies m.E. wenn man auch gleich das QM-System auf Basis MDR und 13485:2016 überarbeitet. Auch dazu gibt es schon eine Veröffentlichung (PD CEN TR 17223_2018 Guidance on the relationship between EN ISO 13485_2016). MfG T. Matz
Bei der 14971:2012 sind für uns als Auditoren vor allem die Änderungen in Kapitel 3 und 4 interessant. Mit dem Prinzip ALARP (as low as reasonably possible) hat man einen guten Weg gefunden um in der gesamten Medizintechnik der Produktsicherheit einen höheren Stellenwert zu geben. Die Gesamtbewertung aller Risiken gemäss Kapitel 4 ist der Punkt in der Norm, der zudem am besten aufzeigt, ob eine Organisation sich seiner Position bewusst ist. Das kann im Markt, gegenüber Kunden oder Mitarbeitern sein. So stellt einerseits die Organisation sicher, dass sie sich selber aber auch ihre Umwelt kennt, sich aber auch gegen innen bewusst ist, welche Herausforderungen anstehen. Es wird also auch dem Fokus des Stakeholdermanagements aus 13485:2016 da bereits Rechnung getragen.
Danke für Ihre wichtigen Gedanken!
Generell halte ich es noch für wichtig, dass man Risiken durch Medizinprodukte für Patienten, Anwender und Dritte von Risiken trennt, denen das (eigene) Unternehmen ausgesetzt ist wie Mitarbeiter, Lieferanten, Kunden, Regularien usw.
Die ISO 9001 hat im Gegensatz zur ISO 13485 die letzteren Risiken explizit mit im Blick. Die ISO 14971:2012 adressiert „nur“ Risiken durch Medizinprodukte.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
wir nutzen in der Entwicklung zur Zeit die EN ISO 14971:2019, um den aktuellen Stand der Technik abzubilden.
Unsere Benannte Stelle fordert von uns nun aber zusätzlich die Einhaltung des Anhangs ZA aus der harmonisierten Norm EN ISO 14971:2012 und eine Synopse zur harmonisierten Fassung.
Wir fragen uns nun, wie wir hier weiter vorgehen. Weitere Nachfragen bei der BS konnten uns hier keine endgültige Klarheiten bieten.
Ist es ausreichend weiterhin nach der 2019 Version zu entwickeln und zusätzlich die Angaben im Anhang ZA 2012 einfließen zu lassen? Oder bedarf es hier noch mehr?
Sehr geehrter Herr Meyer,
danke für Ihre wichtige Frage!
Es ist in der Tat so, dass den internationalen Normen die europäischen Anhänge fehlen. Das ist insbesondere bei der ISO 14971 relevant, weil in diesen Anhängen normative Verschärfungen der internationalen Anforderungen enthalten sind. Daher ist es genauso, wie Sie es sagen optimal:
Die „alten“ Anhänge beachten, aber bereits mit der neueren Norm arbeiten. Die Änderungen sind ja überschaubar, aber z.B. im Fall der Definitionen und der Post-Market Surveillance auch sehr hilfreich.
Beste Grüße, Christian Johner
Sehr geehrte Damen und Herren,
als Hersteller von Medizinprodukten ist es unsere Pflichten ein Risikomanagement in unseres QMS zu implementieren.
Gilt dies doch auf Händler?
müssten wir ein Risikomanagementsystem in unser QMS implementieren, Risikoanalysen , SOPs zum Risikomanagement vorweisen als Händler um ein Gesetzeskonformes QMS vorweisen zu können ?( wir sind Händler ohne Herstellpflichten dh. ohne umverpacken etc .
Mit freundlichen Grüßen,
DORA
Liebe Dora,
tatsächlich gibt es aus dem Medizinprodukte-Kontext erstmal keine direkte regulatorische Anforderung an die Händler-Rolle, Risikomanagement (speziell das nach ISO 14971) durchzuführen.
Wenn Sie aber ein QM-System nach ISO 13485 etabliert haben, dann gibt es dort die Forderung nach einem solchen Risikomanagement-System. Ich habe bereits entsprechende Auditabweichungen bei Kunden gesehen. Es könnte Ihnen auch passieren, dass ein Medizinproduktehersteller per Qualitätssicherungsvereinbarung Forderungen an Sie stellt und dass dabei Risikomanagement verlangt wird (mit der Argumentation, dass der Händler der verlängerte Arm des Herstellers hinsichtlich Aufrechterhaltung der Produkt-Compliance, Kundenbeschwerdemanagement und Meldepflicht gegenüber Behörden ist).
Zusammenfassend: Die MDR/IVDR verlangen von Händlern nicht per se Risikomanagement nach ISO 14971. Eine solche Forderung könnte durch Veränderung der Produkte, Verpackung oder Kennzeichnung erwachsen. Oder aber durch spezifische Forderung, z.B. der ISO 13485 (soweit angewandt) oder anderer Stakeholder.
Herzliche Grüße
Christian Rosenzweig