
Die ISO 24971 ist die Norm, die meine Auditoren-Kollegen oft zücken, wenn es mit Medizinprodukte-Hersteller Diskussionen über die Auslegung der ISO 14971 gibt.
Sie kennen die ISO 24971 nicht? Sie möchten diese Norm nicht kaufen? Kein Problem, ich habe diese Technical Report für Sie gelesen und zusammengefasst.
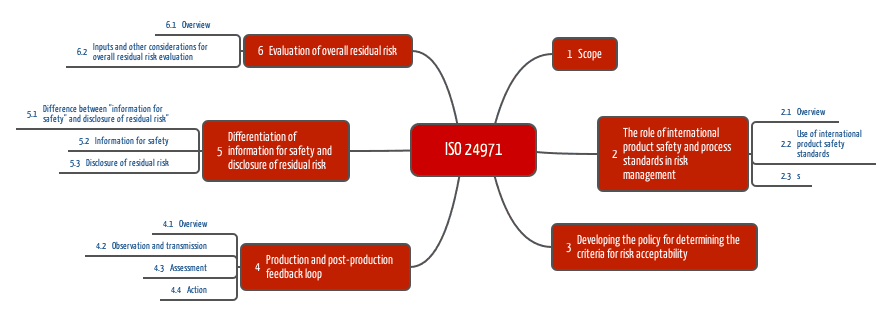
Ziele der ISO 24971
Die Norm möchte bei den folgenden Fragestellungen eine Hilfestellung bieten:
- Wie spielen die verschiedenen Normen mit Bezug zur Sicherheit und zum Risikomanagement zusammen?
- Wie legt man Risikoakzeptanzkriterien fest?
- Welche Informationen aus nachgelagerten Phasen soll man wie nutzen?
- Was ist der Unterschied zwischen Informationen über Restrisiken und Informationen zur sicheren Anwendung?
- Wie bewertet man abschließend das Gesamt-Restrisiko?
Die Kapitel der ISO 24971 spiegeln diese Ziele wider, auf die wir in den folgenden Kapitel eingehen.
Ad 1. Zusammenspiel mit anderen Normen
Die ISO 24971 stellt fest, dass viele Normen wie die Normenfamilie IEC 60601, die IEC 62304, die IEC 62366 und die ISO 10993 (Biologische Bewertung von Medizinprodukten) Risiken und deren Beherrschung adressieren.
Die ISO 24971 beschreibt nun, wie Hersteller vorgehen sollen, um die Forderungen aller Normen zu erfüllen. Beispiele:
- Man soll die Gefährdungen bzw. Gefährdungssituationen mit Hilfe der ISO 14971 identifizieren. Dann soll man diese Liste um die Gefährdungen bzw. Gefährdungssituationen ergänzen, die in den spezifischen Normen zusätzlich genannt werden.
- Wenn die spezifischen Normen konkrete Maßnahmen zur Risikominimierung und konkrete Testverfahren fordern, sind diese umzusetzen.
- Alle Risiken, für die die spezifischen Normen solche konkreten Maßnahmen und Testerfahren fordern, dürfen als akzeptabel eingeschätzt werden, wenn eben diese Maßnahmen umgesetzt und deren Wirksamkeit durch die Testverfahren nachgewiesen wurden.
Ad 2. Festlegung von Risikoakzeptanzkriterien
Die ISO 24971 möchte, dass Hersteller die folgenden Faktoren berücksichtigen, wenn sie Kriterien für die Risikoakzeptanz festlegen:
- Regulatorische Anforderungen – falls existent
- Relevante Normen und Standards (wie die oben genannten)
- Stand der Technik wie er ableitbar ist aus der Fachliteratur und Informationen zu vergleichbaren Medizinprodukten.
- Meinung wichtiger Anspruchsgruppen (z.B. Ärzte, Pflegekräfte, Patienten), wie sie sich in Social Media, Nachrichten, Foren oder den Abteilungen im jeweiligen Haus artikuliert.
Auf die Notwendigkeit, bei der Festlegung der Risikoakzeptanz auch den Nutzen zu betrachten, geht die Norm nicht ein. Das ist sehr bedauerlich.
Ad 3. Informationen aus der nachgelagerten Phase
Der Prozess
Informationen aus den der Entwicklung nachgelagerten Phasen wie Produktion, Installation und Betrieb sollen laut ISO 24971 systematisch
- gesammelt,
- übermittelt und
- bewertet werden und
- notwendige Handlungen auslösen.
Zu berücksichtigende Informationsquellen
Folgende Informationsquellen sollen die Hersteller dabei berücksichtigen:
- Informationen aus der Entwicklung, Produktion beim Hersteller oder bei Dienstleistern
- Informationen von Installation, Wartung, Training
- Informationen der Anwender einschließlich Beschwerden und von Umfragen
- Informationen über Produkte der Mitbewerber wie durch Behördenmeldungen, Fehlerdatenbanken
- Klinische Informationen aus der wissenschaftlichen Fachliteratur oder von eigenen PMCFs (Post-Market Clinical Follow-ups)
- Informationen aus neuen oder überarbeiteten Normen und Richtlinien
- Bei Kombinationsprodukten auch Informationen zum Arzneimittel
Auswertung und Bewertung der Daten
Diese Informationen sollen Hersteller daraufhin bewerten, ob
- die Zweckbestimmung erfüllt und eingehalten ist/wird
- es bisher nicht identifizierte Gefährdungen einschließlich durch Missbrauch gibt
- die Schätzung von Wahrscheinlichkeiten und Schweregrade noch zu stimmen scheint
- die Risikoakzeptanzkriterien und Risiko-Nutzenbewertung noch aktuell sind
Handeln
Abhängig von den Ergebnissen müssen die Hersteller handeln beispielsweise in Form eines Rückrufs oder durch Änderungen in der Entwicklung oder Produktion.
Ad 4. Informationen
Obwohl die ISO 14971 im Anhang J den Unterschied zwischen Informationen über Restrisiken und Informationen zur sicheren Anwendung erklären würde, würden die Hersteller dies dennoch durcheinander bringen – so die Feststellung der ISO 24971. Daher muss sie das nochmals erklären.
Informationen zur sicheren Anwendung
Die Informationen zur sicheren Anwendung von Medizinprodukten dienen als risikominimierende Maßnahmen, deren Wirksamkeit nachzuweisen ist. Ein Beispiel wäre der Hinweis „Vorsicht heiße Oberfläche, nicht berühren!“.
Die ISO 24971 wiederholt, dass diese Maßnahmen die am wenigsten präferierten seien. Zu bevorzugen sind ein inhärent sicheres Design oder (konstruktive) Schutzmaßnahmen.
Informationen über Restrisiken
Informationen über Restrisiken hingegen sind keine risikominimierende Maßnahmen sondern verpflichtende Hinweise an die Anwender mit dem Ziel, Restrisiken und Nebenwirkungen im konkreten Anwendungsfall gegen den Nutzen abwägen zu können.
Ein Beispiel wäre eine strahleninduzierte Krebserkrankung bei einem CT. Dieser Schaden wäre keine Folge einer Fehlfunktion des Geräts oder einer Fehlanwendung. Diese Information wird das Risiko dieser Nebenwirkung auch nicht minimieren.
Ad 5. Bewertung der Akzeptanz des Gesamt-Restrisikos
ISO 24971 zu den Schwierigkeiten
Die ISO 24971 benennt die Schwierigkeiten, auf die Hersteller stoßen, wenn sie das Gesamt-Restrisiko auf Akzeptanz bewerten müssen:
- Man kann nicht mathematisch aus den Einzelrisiken und deren Akzeptanz ein Gesamt-Restrisiko und dessen Akzeptanz ableiten.
- Die Abschätzung von Wahrscheinlichkeiten und Schweregraden der Einzelrisiken ist mit einer hohen Unsicherheit behaftet.
- Es ist unklar, ob die Akzeptanzkriterien für die Einzelrisiken den Akzeptanzkriterien für das Gesamtrisiko gleichen müssen. Die ISO 14971 fordert das nicht.
Hinweise der ISO 24971, wie man die Akzeptanz des Gesamt-Restrisikos bewerten kann
Eins vorweg: Die ultimative Antwort auf die Frage, wann ein Gesamt-Restrisiko als akzeptabel bewertet werden darf, gibt die ISO 24971 nicht. Aber sie gibt uns zumindest Hinweise:
- Eine notwendige Voraussetzung ist, dass alle festgelegten Maßnahmen umgesetzt und deren Wirksamkeit nachgewiesen wurden.
- Wenn das Medizinprodukt bei einem gleichen oder höheren Nutzen ein gleiches oder niedrigeres Risiko als ein vergleichbares Produkt hat, ist das ein Hinweis, dass dieses Risiko (Gesamt-Restrisiko) akzeptabel ist.
- Falls in einer grafischen Darstellung viele Einzelrisiken nur nahe unterhalb der Akzeptanzschwelle liegen, kann das ein Hinweis darauf sein, dass das Gesamtrisiko oberhalb liegt.
- Muss der Hersteller beim Design zwischen verschiedenen Risiken abwägen (und das kleinere Übel wählen), so mag ihm dies ein Hinweis sein, dass er noch nicht das optimale Design gefunden haben, d.h. das Design mit minimalen Restrisiken.
Die Norm hat noch weitere Tipps für uns parat:
- Wir sollen auch externe Experten einbeziehen.
- Wir sollen die Ergebnisse der klinischen Bewertung und der Usability Bewertung mit einbeziehen (Anmerkung: das ist nicht nur ein Tipp, sondern normativ gefordert!)
- Schlussendlich basiert die Bewertung des Gesamt-Restrisikos auf einer klinischen Entscheidung.
ISO 24971, ein Fazit
Die ISO 24971 gibt zweifelsohne nützliche Hinweise zur Anwendung des Risikomanagements – zumindest mit Bezug auf die fünf oben genannten Themen. Leider schlägt sie keine konkreten Methoden vor, sie erklärt eher, nennt Beispiele, könnte aber an mancher Stelle noch mehr handlungsleitend sein.
Die Norm ist mit 20 Seiten erfrischend kurz. Allerdings bezieht sie sich auf die ISO 14971:2007. Eine Interpretation der unsäglichen oder zumindest unglücklichen Z-Anhänge der ISO EN 14971:2012 und der darin aufgeworfenen Probleme liefert die ISO 24971 leider nicht.
Dennoch ist die Norm lesenswert – und sei es nur in Form dieser Zusammenfassung :-).

Sehr geehrter Herr Johner,
ist Ihnen bekannt, ob zur DIN EN ISO 14971:2020-07 die vorliegende Norm ISO/TR 24971:2020-06 auch ins deutsche übersetzt wird bzw. als DIN EN Norm erscheinen wird?
Kann vom Grundsatz her die ISO/TR 24971:2020-0 auch für die DIN EN ISO 14971:2020-07 genutzt werden?
Herzlichen Gruß
Franjo Peters
Lieber Herr Peters,
ob die ISO/TR 24971:2020-06 noch ins Deutsche übersetzt wird, kann ich Ihnen leider nicht sagen. Weder der Beuth-Verlag, noch das DIN listen einen entsprechenden Entwurf.
Da die DIN EN ISO 14971:2020-07 im Grunde die deutsche Übersetzung der internationalen Version ist, können Sie die ISO/TR 24971:2020 darauf anwenden.
Herzliche Grüße
Christian Rosenzweig