
Die Prozess-FMEA (pFMEA) ist eine Methode zur systematischen Analyse von Risiken, die sich durch Fehler in Prozessen wie der Produktion und Reinigung von Produkten ergeben.
Gesetze wie die MDR und Normen wie die ISO 13485 verpflichten die Hersteller von Medizinprodukten dazu, solche Prozessrisiken zu identifizieren und zu beherrschen.
1. Was ist die pFMEA?
a) Die pFMEA ist eine Methode, um Auswirkungen von Prozessfehlern zu finden
Die Prozess-FMEA (pFMEA) ist wie die Design-Analyse (dFMEA) eine Ausprägung der FMEA, der Failure Mode and Effect Analysis. Damit ist sie ein Bottom-up-Verfahren, das zu angenommenen Fehlern (Failure Modes) die unbekannten (negativen) Auswirkungen (Effects) sucht (analysiert).
| Prozess | Fehler im Prozess (Beispiel) | Negative Auswirkung auf das Prozessergebnis (Beispiel) |
| Dampfsterilisation | Temperatur ist nicht hoch genug | Gegenstand ist nicht steril |
| Leiterplattenbestückung | Falscher Gurt mit Elektronikbauteilen eingelegt | Leiterplatte ist fehlerhaft bestückt, Elektronik funktioniert nicht wie vorgesehen |
| Post-Market Surveillance | Verantwortliche Person bzw. Bot finden Informationsquelle nicht | Bericht (z. B. PSUR) ist unvollständig |
| Statische Code-Analyse | Konfigurationsdatei des Analysewerkzeugs wird überschrieben | Testbericht warnt nicht, dass die zyklomatische Komplexität des Codes zu hoch ist |
b) Die pFMEA ist (meist) keine Methode zur Risikoanalyse
Im Kontext der Medizinprodukte sind die unbekannten und meist unerwünschten Folgen bzw. Auswirkungen (Effects) dieser Fehler Gefährdungen, Gefährdungssituationen und Schäden. Kennt man die Wahrscheinlichkeit und den Schweregrad dieser resultierenden Schäden, kennt man auch die Risiken.
Deshalb wird die FMEA vornehmlich auch als Methode zur Risikoanalyse verstanden. Allerdings ist das nicht ganz zutreffend, da die Methode nur bedingt geeignet ist, um die Schweregrade von Schäden und Wahrscheinlichkeiten der Schäden zu bestimmen.
Zudem sind die Auswirkungen keine Schäden im Sinne der ISO 14971 (z. B. physische Beeinträchtigungen von Patienten). Vielmehr handelt es sich um Elemente einer Ursachenkette, die erst im weiteren Verlauf zu diesen Schäden führt.
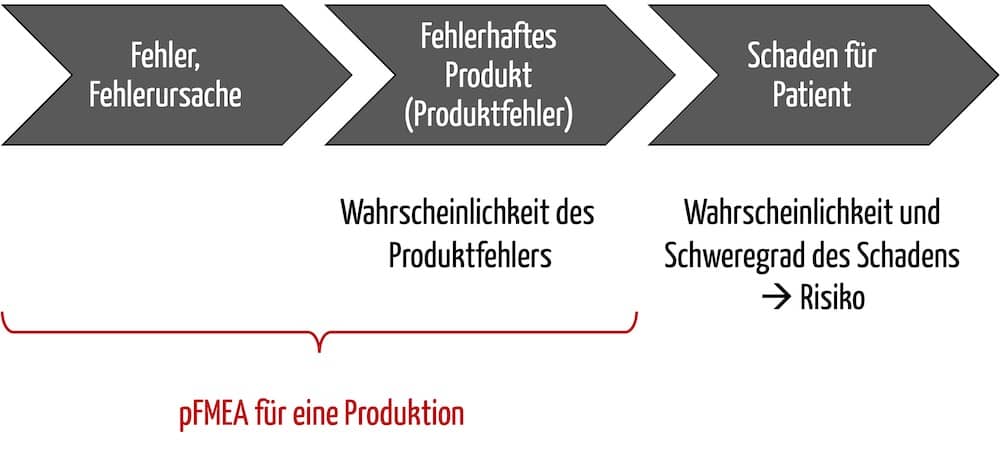
Beispielsweise ist ein nicht steriles Produkt (siehe Tabelle 1) kein Schaden, sondern eine mögliche Schadensquelle und damit definitionsgemäß eine Gefährdung und kein Risiko.
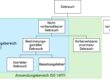
c) Die pFMEA ist eine gute Ergänzung zur dFMEA
Wie die Akronyme bereits nahelegen, untersucht die pFMEA die Auswirkungen fehlerhafter Prozesse. Die dFMEA hat hingegen die Auswirkungen fehlerhafter Produkte im Fokus. Das „d“ in dFMEA bezieht sich auf das Design; Design hier im Sinne des Entwurfs und der Konstruktion von Produkten, weniger im Sinne des grafischen Designs.
Das Ergebnis einer pFMEA besteht in der Abschätzung der Auswirkungen eines Prozessfehlers auf das Ergebnis. So ein (unerwünschtes) Prozessergebnis kann beispielsweise eine nicht spezifikationsgemäß produzierte Komponente sein. Diese nicht spezifikationsgemäße Komponente ist der Startpunkt der dFMEA. Die Methode untersucht die Auswirkungen von nicht spezifikationsgemäßen Komponenten auf das gesamte Produkt und letztlich auf die Patienten.
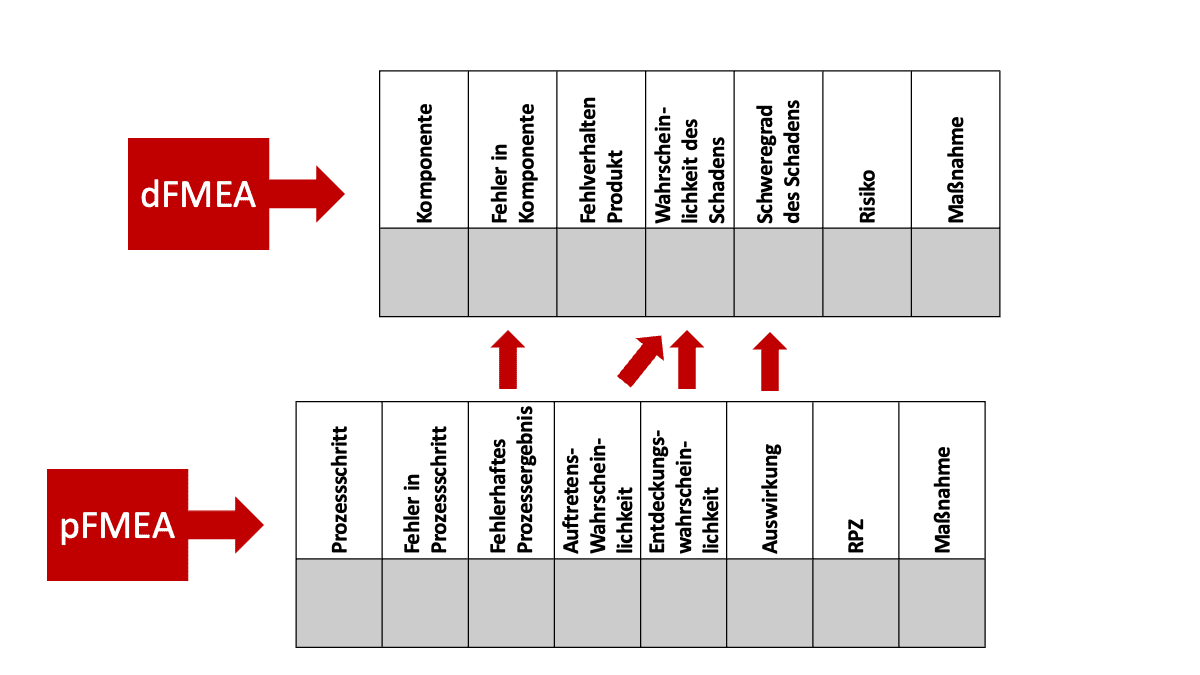
Damit kann die pFMEA der dFMEA als Input dienen, indem sie wesentliche Informationen liefert:
- Art des nicht spezifikationsgemäßen Verhaltens (z. B. fehlerhaft bestückte Platine)
- Ausmaß des nicht spezifikationsgemäßen Verhaltens (z. B. Anzahl der verbleibenden Keime auf einem sterilisierten Teil des Medizinprodukts)
- Wahrscheinlichkeit des nicht spezifikationsgemäßen Verhaltens
2. Wann sollte die pFMEA zum Einsatz kommen?
a) pFMEA beim Entwurf neuer Prozesse
Nicht nur bei Kernprozessen
Die pFMEA sollte immer dann zum Einsatz kommen, wenn eine Organisation neue Prozesse etabliert. Das betrifft nicht nur Kernprozesse wie Entwicklung, Produktion und Wartung von Produkten, sondern auch Unterstützungsprozesse wie:
- Einkauf
- Lieferung, Logistik, Lagerung
- Überwachung neuer und geänderter Regularien
- Post-Market Surveillance
- Vertrieb
- Schulungen, Training von Mitarbeitenden und von Kunden
Ggf. mehr als eine pFMEA pro Prozess
Bei den Kernprozessen sollte die Organisation nicht nur ein einzelne pFMEA durchführen. Vielmehr kommen im Rahmen dieser Prozesse mehrere Subprozesse bzw. Verfahren zur Anwendung, die einzeln analysiert werden sollten, wie die folgenden Beispiele zeigen:
- Entwicklung
- Modellierung und Simulation von physikalischen Eigenschaften, z. B. der mechanischen Festigkeit
- Testen von Software und statische Code-Analyse
- Sammeln von Trainingsdaten und Trainieren von Modellen beim maschinellen Lernen (Künstliche Intelligenz)
- Produktion
- Identifikation und Rückverfolgbarkeit von Bauteilen und Materialien
- Bestückung von Leiterplatten
- Montage von Komponenten
- Sterilisation von Produkten
- Verpackung und Labeling von Produkten
pFMEA zur Prozessgestaltung
Die pFMEA hat zum Ziel, unerwünschte und unbekannte Auswirkungen fehlerhafter Prozesse zu analysieren. Damit hilft sie, notwendige Gegenmaßnahmen wie zusätzliche Prüfschritte zu bestimmen. Und das wiederum führt zu Änderungen der Prozesse: Das Prozessdesign, die pFMEA und die Prozessänderung erfolgen iterativ.
b) pFMEA bei der Änderung bestehender Prozesse
Es gibt viele Anlässe, aufgrund derer Prozesse geändert werden müssen:
- Es sind bisher unbekannte Risiken aufgetreten.
- Bekannte Risiken wurden falsch (zu niedrig) eingeschätzt.
- Prozess-Inputs ändern sich, weil z. B. Bauteile nicht mehr verfügbar sind.
- Prozesselemente müssen geändert werden: Es erfolgt z. B. ein Software-Update der Steuerelektronik für eine Produktionsstraße.
- Die Prozess-Outputs müssen geändert werden.
- Betriebswirtschaftliche Gründe zwingen den Hersteller zu einer höheren Automatisierung von Prozessen.
Diese Änderungen muss die Organisation daraufhin bewerten, ob
- neue (unbekannte) Auswirkungen oder
- bekannte Auswirkungen mit einer anderen Wahrscheinlichkeit oder
- bekannte Auswirkungen mit einem anderen Schweregrad
die Folgen sein können. Genau dafür ist die pFMEA prädestiniert.
c) pFMEA zur Änderung bestehender Prozesse
Die pFMEA kann nicht nur die notwendige Folge einer Prozessänderung sein, sondern auch selbst eine Änderung auslösen. Denn die Organisation ist mithilfe der pFMEA in der Lage, Schwachstellen in den Prozessen zu identifizieren und diese zu verbessern, selbst wenn keine negativen Auswirkungen auf Patienten, Anwender und Dritte die Folge wären.
Mögliche Schwachstellen sind:
- Die Anzahl unkritischer Fehler
- Die Notwendigkeit von weiteren Arbeits- und Prüfschritten
- Die Durchlaufdauer des Prozesses
- Der Verbrauch an Energie und Materialien
- Die Abgrenzung von und das Zusammenspiel mit anderen Prozessen
- Die Entscheidung für oder gegen ein Outsourcing
d) pFMEA bei der Vorbereitung der Prozessvalidierung
Die regulatorischen Vorgaben (siehe Kapitel 4 dieses Artikels) zwingen die Hersteller, die Prozesse zu validieren. Diese Validierung bedeutet für Hersteller hohe Aufwände. So sind eine vollständige Prüfung aller Prozessparameter oder das vollständige Testen einer Prozesssoftware meistens sogar unmöglich.
Daher erlauben die gesetzlichen und normativen Vorgaben einen risikobasierten Ansatz. Das heißt, dass die Hersteller die Aufwände für die Validierung des Prozesses an die Risiken anpassen dürfen, die aus diesem Prozess hervorgehen.
Die pFMEA trägt dazu bei, diese Risiken zu quantifizieren, auch wenn sie selbst keine Methode zur Risikoanalyse im Sinne der ISO 14971 ist.
In diesem Artikel zur Prozessvalidierung erfahren Sie mehr zum Vorgehen und zu den regulatorischen Anforderungen, die Sie dabei erfüllen müssen.
3. Wie läuft eine pFMEA ab?
Schritt 1: Planung
Bei der Planung der pFMEA legt die Organisation die folgenden Aspekte fest:
- Prozess, der analysiert werden soll
- Team, das die pFMEA durchführen oder daran beteiligt werden soll
- Dafür notwendige Aktivitäten (werden im Folgenden beschrieben)
- Zeitpunkt und Zeitdauer für die pFMEA
- Regulatorische Anforderungen, die erfüllt werden müssen
- Ggf. Methoden und Werkzeuge für z. B. die Dokumentation
Schritt 2: Vorbereitung
Bei der Vorbereitung der pFMEA sichtet und ergänzt das Team die Prozessbeschreibung. Fehlt diese Beschreibung, so holt das Team diese Dokumentation nach.
Die Prozessbeschreibung sollte umfassen:
- Prozessschritte. Für jeden Schritt
- die Inputs (Materialien, Energien, Informationen)
- die Outputs (Materialien, Energien, Informationen)
- die daran beteiligten Personen
- die vorgesehenen Aktivitäten ggf. mit zugehörigen Arbeitsanweisungen und Anforderungen
- die daran beteiligten Systeme (Werkzeuge, Maschinen, Software usw.) ggf. mit vorhandenen Validierungsaufzeichnungen
- Anforderungen an das Prozessergebnis
- Für jedes Prozessergebnis eine Abschätzung der Folgen, wenn dieses nicht erfüllt ist. Als Vereinfachung kann jedes Prozessergebnis als kritisch oder nicht kritisch klassifiziert werden. Diese Klassifizierung lässt sich z. B. aus einer dFMEA ableiten.
Um diese Prozesse übersichtlich zu dokumentieren, eignen sich Diagramme, die z. B. der Spezifikationssprache Business Process Modeling Notation (BPMN) erstellt werden.
Schritt 3: Durchführung
Die eigentliche pFMEA besteht darin, für jeden Prozessschritt zu überlegen, welche Fehler auftreten können und welche Folgen diese für das Prozessergebnis hätten. Dabei unterstützen die folgenden Leitfragen:
- Was sind die Folgen für den Output eines Arbeitsschritts,
- wenn dessen Inputs nicht der Spezifikation genügen?
- wenn dessen Aktivitäten nicht oder nicht wie spezifiziert durchgeführt werden?
- wenn sich ein System (Maschine, Werkzeuge usw.) nicht spezifikationsgemäß verhält?
- Was sind die Folgen, wenn ein Arbeitsschritt nicht durchgeführt wird oder zwei Arbeitsschritte vertauscht werden?
- Wie erkennt man, dass einer der oben beschriebenen Fehler aufgetreten ist?
Die Ishikawa-Methode hilft Ihnen, mögliche Fehlerquellen für jeden Prozessschritt zu identifizieren. Die HAZOP-Methode (IEC 61882) ist sehr nützlich, um die verschiedenen Formen fehlerhafter Inputs und Outputs zu beschreiben.
Die Ergebnisse der pFMEA müssen dokumentiert werden. Dafür eignet sich eine tabellarische Form:
| Prozessschritt | Fehler beim Prozessschritt | Ggf. Ursache für den Fehler | Wahrscheinlichkeit für den Fehler | Entdeckungswahrscheinlichkeit für den Fehler | Auswirkung des Fehlers | RPZ | Maßnahmen |
| 1: … | |||||||
| 2: … |
In der pFMEA werden drei Größen quantitativ erfasst und jeweils mit Werten zwischen 0 und 10 versehen:
- Wahrscheinlichkeit für den Fehler
- Eins minus Entdeckungswahrscheinlichkeit, wobei die Entdeckungswahrscheinlichkeit einen Wert zwischen 0 (sichere Entdeckung) und 1 (Entdeckung unmöglich) einnimmt. Wenn die Entdeckungswahrscheinlichkeit 1 (d. h. 100 Prozent) ist, ist der Wert null.
- Auswirkung des Fehlers (0 = keine Auswirkung, 10 = maximal schwere Auswirkung)
Die Risikoprioritätszahl (RPZ) ist das Produkt dieser drei quantitativen Größen und kann Werte zwischen 0 und 1.000 annehmen.
Viele Firmen nutzen die Zahl 125 als Grenzwert, unterhalb dessen keine risikominimierenden Maßnahmen notwendig sind. Die ISO 14971 und die MDR bzw. IVDR bestehen allerdings darauf, die Risiken so weit wie möglich zu reduzieren. Daher müsste diese Zahl zumindest begründet werden können. Besser wäre es, darauf zu verzichten und für jedes „Risiko“ einzeln zu entscheiden, ob Maßnahmen notwendig sind.
Schritt 4: Ergebnisbewertung
Die Risikoprioritätszahl gibt einen Hinweis darauf, wie folgenreich ein Fehler ist. Allerdings wird sie inzwischen nicht mehr als ein Maß für das Risiko angesehen, sondern als Hilfsmittel für die Priorisierung von Aufgaben (insbesondere Maßnahmen).
Die finale Bewertung des Risikos im Sinne der ISO 14971 obliegt nicht mehr (alleine) dem pFMEA-Team. Vielmehr müssen die Folgen fehlerhafter Prozess-Outputs bis hin zu Patienten, Anwendern und Dritten extrapoliert werden.
Schritt 5: Ergebnisweitergabe
Als wesentliches Ergebnis der pFMEA liegt die oben gezeigte Tabelle 2 vor. Die Maßnahmen sind nicht Bestandteil dieser Analyse. Die Spalte „Maßnahmen“ hat sich allerdings bewährt, um sicherzustellen, dass zu den identifizierten Risiken auch tatsächlich Maßnahmen ergriffen wurden.
4. Welche Fehler sollten bei der pFMEA vermieden werden?
Fehler 1: Ziele unklar
Das Team sollte ein gemeinsames Verständnis der Ziele haben. Das betrifft:
- „Scope“, insbesondere den Prozess (Ist z. B. die ganze Produktion gemeint oder „nur“ die Montage?)
- Zielsetzung (Geht es z. B. um das Erfüllen regulatorischer Anforderungen oder um eine Verbesserung des Prozesses?)
- Bedeutung der pFMEA (Wären Patientenschäden die möglichen Folgen einer insuffizienten pFMEA oder „nur“ ein verlangsamter Prozess?)
- Regulatorische Anforderungen, die damit oder dabei erfüllt werden müssen (z. B. die der FDA oder die der EU?)
Fehler 2: Team falsch zusammengesetzt
Im Team sollten die folgenden Rollen zumindest zeitweise beteiligt sein:
- Risikomanager
- beherrscht die Methodik
- kennt Auswirkungen eines suboptimalen Prozessergebnisses auf Patienten, Anwender und Dritte
- kennt regulatorische Anforderungen
- Prozessverantwortliche
- kennt den Prozess
- Am Prozess beteiligte Personen
- wissen, was schiefgehen kann
- Qualitätsmanager
Fehler 3: Zeitpunkt ist falsch gewählt
Die pFMEA sollte zum Einsatz kommen:
- Wenn der Prozess erstmalig entworfen wird
- Falls eine Rückmeldung aus nachgelagerten Phasen dazu Anlass gibt
- Wenn der Prozess einschließlich der daran beteiligten Systeme geändert werden soll
- Regelmäßig, um auch den State of the Art zu überprüfen
Fehler 4: Abstimmung fehlt
Die pFMEA alleine liefert nicht die gewünschten Informationen, um Risiken durch Medizinprodukte zu identifizieren und zu quantifizieren. Vielmehr müssen betrachtet werden:
- Alle Prozesse, die Inputs für den untersuchten Prozess liefern (z. B. die Komponentenproduktion für die Montage)
- Alle Prozesse, die mit den Outputs des Prozesses arbeiten (z. B. die Produktion mit den Ergebnissen der Entwicklung)
- Das gesamte Risikomanagement, das sowohl die Entwicklung (dFMEA) als auch die Produktion sowie nachgelagerte Prozesse (pFMEA) umfassen muss
Damit bedarf es oft einer Abstimmung mehrerer Prozessverantwortlicher, selbst wenn nur ein Prozess geändert wird.
Fehler 5: RPZ und ihre Faktoren werden missverstanden
Die Risikoprioritätszahl ist kein Maß für das Risiko gemäß ISO 14971. Auch darf der Faktor „Auswirkung“ nicht mit dem Schweregrad von Schäden nach ISO 14971 verwechselt werden.
Die Multiplikation von drei numerischen Werten erzeugt oft die falsche Illusion einer Genauigkeit. Aber meist ist jeder der Faktoren nur das Ergebnis einer Schätzung, und die Multiplikation kann die Ungenauigkeit vergrößern.
In unserem Beitrag zur Risikoprioritätszahl werden diese Überlegungen detailliert dargelegt.
5.Welche regulatorischen Anforderungen hilft die pFMEA zu erfüllen?
Hersteller müssen nach Stand der Technik die Risiken durch ihre Medizinprodukte identifizieren und beherrschen. Die pFMEA ist eine Methode, die diesem Stand der Technik entspricht.
Diese Anforderungen finden sich u. a. in den folgenden regulatorischen Vorgaben:
- MDR, IVDR
- Artikel 10 (2)
- Anhang I (3)
- Anhang I in vielen weiteren Abschnitten, u. a. (10), (11)
- Anhang II Abschnitt (3), (5)
- ISO 13485
- Vorgaben zum risikobasierten Ansatz, u. a. beim Validieren von Prozessen, Messmitteln und computerisierter Software
- Vorgaben zur Prozessvalidierung, u. a. Kapitel 7.5.6
- FDA 21 CFR part 820 (der weitgehend durch die ISO 13485 abgelöst wird/wurde)
Lesen Sie in diesem Beitrag zur Computerized Software Validation, die ebenfalls risikobasiert erfolgen soll oder kann, warum die pFMEA eine wertvolle Methode ist.
6. Fazit und Zusammenfassung
Die pFMEA dient der systematischen Analyse der Auswirkungen, die ein Fehler innerhalb eines Prozesses nach sich ziehen kann.
Einige Auditoren betrachten die pFMEA als Stand der Technik. Unabhängig davon sollten Organisationen die pFMEA als Methode in Betracht ziehen, insbesondere bei Prozessen, die eine Auswirkung auf die Konformität der Produkte haben, z. B. wie die Produktion.
Mit der pFMEA schaffen die Organisationen nicht nur Konformität, sondern sie können ihre Aufwände für z. B. Produktprüfungen risikobasiert gestalten und dadurch unnötige Aufwände vermeiden.
Das Johner Institut unterstützt Medizinproduktehersteller und deren Dienstleister beim Aufbau von QM-Systemen und bei der Risikoanalyse für Produkte und Prozesse. Treten Sie mit uns in Kontakt.
Im Auditgarant stellt das Johner Institut mehrere Videoserien und dazu passende Templates bereit, mit denen Hersteller eine dFMEA oder pFMEA gesetzeskonform durchführen können.
Änderungshistorie
- 2025-05-14: Nur kleine redaktionelle Änderungen, v. a. die Info-Kästen neu gestaltet, Links ergänzt, Rechtschreibfehler korrigiert
- 2022-05-03: Artikel in jetziger Form veröffentlicht
Sehr geehrter Prof. Johner,
wieder mal ein sehr interessanter Artikel.
Die obere Abbildung in 1c) suggeriert, dass sich mit der dFMEA die Wahrscheinlichkeit und der Schweregrad des Schadens und somit das Risiko ermitteln lässt. Dem ist aber nicht so. Analysiert werden, wie in der pFMEA auch, mögliche Auswirkungen eines Fehlers, also maximal bis zu dem Punkt „Fehlverhalten Produkt“. Alles andere ist Risikoanalyse.
Schöne Grüsse
Sehr geehrter Herr Stegmaier,
das ist ein sehr wertvoller Hinweis!
Wir beobachten, dass die FMEA bei vielen Medizinprodukteherstellern (so auch suggeriert durch die ISO 14971) als Methode zur Risikoanalyse genutzt wird d.h. dass die Ursachenkette damit bis zum Patienten nachverfolgt wird.
Im Automobilbau hingegen gibt es fast immer die von Ihnen angesprochene Trennung von FMEA und Risikoanalyse.
Eine Zerlegung der Ursachenketten halten wir generell für sinnvoll. Aber ich wollte der Tatsache Rechnung tragen, dass die Medizinproduktehersteller meist nur eine „Risikotabelle“ verwenden (die sie bezeichnenderweise oft als FMEA-Tabelle bezeichnen).
Nochmals besten Dank für Ihren sehr relevanten Input!
Viele Grüße
Christian Johner
M.E. ist eine RPZ, die einfach nur aus dem Produkt der genannten 3 Größen besteht, nicht mehr Stand der Technik. Deshalb ist die Automobilindustrie auch davon abgekommen. Wenn man weiter eine RPZ verwendet, greift man besser zu alternativen Berechnungsmethoden. Es wäre wahrscheinlich sinvoll, die RPZ auch im Artikel noch etwas kritischer zu beleuchten und das einfache Produkt nur als (eher schlechtes) Beispiel aufzuführen.
Sehr geehrter Herr Müller,
danke für den Hinweis! Sie haben Recht, der Stand der Technik hat sich weiterentwickelt.
Unsere Expertise beschränkt sich auf die Medizinproduktewelt. Falls Sie Best Practices im Automobilbau beitragen könnten, wäre das großartig.
Nochmals besten Dank!
Viele Grüße
Christian Johner