
| Inhaltsübersicht |
|---|
| Anforderungen der ISO 14971 » |
| Welche Restrisiken sind akzeptabel? » |
| Zahlen, die Sie kennen sollten » |
| Informationen über Restrisiken » |
Lesen Sie hier, wie Sie zu belastbaren Akzeptanzkriterien für das Restrisiko kommen. Diese Zahlen können Ihnen nützlich sein.
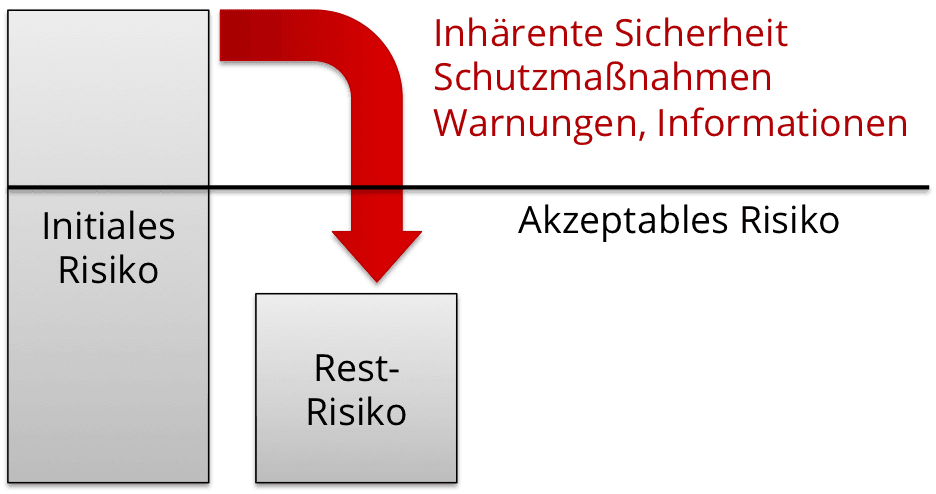
Restrisiko: Diesbezügliche Anforderungen der ISO 14971
Die ISO 14971 fordert von den Herstellern, jedes Restrisiko auf Akzeptanz zu beurteilen. Diese Bewertung muss anhand der im Risikomanagementplan festgelegten Kriterien erfolgen. Die Hersteller sind verpflichtet sicherzustellen, dass der Nutzen des Produkts die Restrisiken überwiegt.
Weiter müssen die Hersteller darüber entscheiden, welche Restrisiken er z.B. in einer Gebrauchsanweisung offenlegt.
Schließlich fordert die Norm, dass die Restrisiken und deren Akzeptanz solange kontinuierlich weiter bewerten, wie die Produkte im Markt sind.
Welche Restrisiken sind akzeptabel?
Ein ausführlicher Beitrag zur Risikoakzeptanz diskutiert, dass ein Risiko nur dann akzeptabel sein kann, wenn der Nutzen höher ist.
Beispiel 1: Restrisiko bei Produkten mit potenziell tödlichen Folgen
Ein Bestrahlungsgerät dient v.a. dazu, an Krebs erkrankte Patienten zu behandeln. Die Strahlung verursacht aber Strahlenschäden, die wieder neuen Krebs auslösen können.
Das Risiko dieser strahlungsinduzierten potenziell tödlichen Erkrankung ist nur dann akzeptabel, wenn der Nutzen höher ist. Konkret muss die Anzahl der Toten, die mit dem Gerät auftreten, kleiner sein als die Anzahl der Toten, die ohne das Gerät auftreten. Mit „ohne Gerät“ ist aber nicht gemeint, dass die Patienten überhaupt nicht behandelt würden, sondern mit der besten Alternative z.B. einem anderen Bestrahlungsgerät, einer Chemotherapie, einer OP usw.
Diese Alternativen stellen den Stand der Technik dar.
Beispiel 2: Restrisiko bei Produkten ohne potenziell tödliche Folgen
Bei Medizinprodukten, die nicht lebensrettend sein können, greift diese Argumentation nicht. Ein Beispiel wäre ein Laufband für die Rehabilitation. Im besten Fall kann das Produkt dazu beitragen, dass die Patienten nach einer Erkrankung oder einem Unfall schneller wieder auf den Füßen sind – im wahrsten Sinne des Worts.
Dennoch können diese Produkte den Tod von Patienten verursachen:
- Der Patient stürzt, beispielsweise weil das Laufband blockiert, und schlägt mit dem Kopf auf dem Boden auf.
- Die Isolation eines Kabels bricht, daher liegt die Netzspannung direkt auf dem Gehäuse, und der Patient erleidet einen Stromschlag.
Für die Abwägung, ob ein Restrisiko akzeptabel ist, greift der Vergleich der Anzahl von Toten wie im ersten Beispiel nicht mehr. Was tun?
Mögliche Risikoakzeptanzkriterien
Um entscheiden zu können, ob ein Restrisiko akzeptabel ist, bieten sich folgende Typen an Kriterien an:
- Direkter Vergleich des Nutzens und der Risiken des Produkts und der besten Alternative z.B. anhand
- Anzahl der Toten
- Anzahl der Verletzten
- Lebensdauer
- Stand der Technik: Im zweiten Beispiel würde man das Restrisiko durch einen Stromschlag akzeptieren, wenn die elektrische Sicherheit dem Stand der Technik entspricht. D.h. konkret, wenn das Medizinprodukt konform mit der IEC 60601-1 entwickelt und produziert wurde.
- Vergleich mit den normen Risiken des Lebens: In anderen Branchen wie der Luftfahrtindustrie vergleicht man die Risiken mit denen des normalen Lebens. Nur wenn letztere Risiken deutlich (z.B. mindestens eine Größenordnung) höher sind, bewertet man ein Restrisiko als akzeptabel.
Restrisiken: Zahlen, die Sie kennen sollten
Die folgenden Zahlen können Ihnen dienlich sein, wenn Sie über die Akzeptanz eines Restrisikos entscheiden müssen:
- In deutschen Krankenhäusern treten pro Jahr ca. 500.000 Infektionen (z.B. Wundinfektionen nach Operationen) auf [Quelle: Charité]. Ärzte, die behaupten, durch ein Medizinprodukt dürften überhaupt keine Todesfälle verursacht werden, kann man darauf hinweisen, dass zwischen 80.000 und 180.000 Infektionen vermeidbar wären [Quelle].
- 3000 Patienten sterben jährlich im Krankenhaus durch MRSA-Infektionen, davon wäre ein Dritte vermeidbar [Quelle]. Die Wahrscheinlichkeit an so einem Keim zu sterben beträgt somit ca. ein Promille.
- Die ISO 13849-1 fordert, dass Maschinenanwender seltener als jede10E-7-te Arbeitsstunde einem Ereignis mit schwerwiegender Folge ausgesetzt sein darf.
- Die Wahrscheinlichkeit, durch einen Autounfall ums Leben zu kommen beträgt ca. 2x10E-7 pro Fahrstunde oder 2,7 Todesfälle pro Milliarde Personenkilometer [Quelle].
- Die geringste erfasste Sterberate haben ca. 15 jährige Mädchen: 3,5E-8/h [Quelle S. 130. Lesenswert ist das Kapitel 5.2]
Diese Zahlen helfen Ihnen, das Restrisiko mit den Risiken des normalen Lebens bzw. den Risiken in einem Krankenhaus zu vergleichen.
Soll ich über Restrisiken informieren?
Darf man oder soll man oder muss man Benutzer über Restrisiken informieren? Immer wieder bekomme ich – für mich fast überraschend – entsprechende Fragen in meiner Auditsprechstunde zu hören. Den Grund für diese Irritation liefert die ISO 14971 selbst. Diese besagt:
Wenn es dagegen die Information des Anwenders über das verbleibende Restrisiko (nach Abschluss der Risikobeherrschung) gemäß Abschnitt 6.4 und Anhang J.3 ist, hat dies keine weiteren Auswirkungen auf das Risiko, insbesondere keine Verminderung.
Hier sollten Sie zwei Dinge unterscheiden:
- Es gibt Warnungen und Hinweise, die eine Anleitung zur Vermeidung von Schäden sein sollen (risikominimierend).
- Es gibt Hinweise, die nicht risikominimierend sind, sondern nur den Anwender über Restrisiken „nur“ informieren. Die Hersteller müssen entscheidend, welche Restrisiken sie offenlegen (s.o.).
Der erste Punkt ist „offiziell“ nicht mehr erlaubt. D.h. Sie müssen über Restrisiken informieren, Sie können aber Informationen nicht mehr als risikominimierende Maßnahmen nutzen. So will es Brüssel. Dieser Meinung widersprechen aber die meisten benannten Stellen. Zu Recht:
Ein Warnhinweis bei einer Software, der beispielsweise erscheint, wenn jemand ein Mamma Carcinom bei einem Mann eingibt und vorschlägt das Geschlecht bzw. die Diagnose nochmals zu prüfen, kann durchaus wirksam sein. Solch ein Hinweis wäre nicht nur eine Information über ein Restrisiko, sondern tatsächlich eine risikominimierende Maßnahme.
Dass ein Hinweis in einer Gebrauchsanweisung kaum als risikominimierende Maßnahme taugt, macht folgende Überlegung klar:
Eine typische Risikoakzeptanzmatrix hat Wahrscheinlichkeitskategorien, die zwei Größenordnungen umfassen. Ein Hinweis in der Gebrauchsanweisung führt meist zu keiner Reduktion der Wahrscheinlichkeitsklasse (d.h. um 99%). Das war schon immer so. Das relativiert auch die ganze Diskussion, die die ISO 14971 losgetreten hat.

Guten Morgen in die Runde,
ich bitte um Vorsicht an einer wichtigen Stelle.
Der Anhang ZA (ZB, ZC) und die Norm sollten nicht gleich gesetzt werden. Diese Anhänge sind von einer anderen Gruppe „erarbeitet“ worden, die Wortlaut und Intention der Norm nicht an allen Stellen vollständig verstanden haben. Siehe hierzu auch die Stellungnahme von Prof. Dr. Schwanbom im MPJ, 20. Jahrgang, Heft 4, 2013, speziell S. 264.
Die „…Warnungen und Hinweise, die eine Anleitung zur Vermeidung von Schäden sein sollen…“ sind keineswegs nicht mehr erlaubt! Vielmehr glaubt die Kommission, dass solche Hinweise keine das Risiko reduzierende Wirkung haben dürften. Das ist so nicht richtig, basiert die „inhaltliche Abweichung 7“ des Anhang ZA doch auf einem Lesefehler.
Nach wie vor haben Sicherheitswarnungen die Möglichkeit, Risiken zu senken, wenn sie konkrete Handlungsanweisungen oder Fachinformationen (z.B. „nicht re-sterilisieren“) beinhalten. Allein, die reine Information über Restrisiken senkt diese nicht weiter, das sollte jedem klar sein.
Ich wünsche mir, dass Normen und die Kommission uns nicht (weiter?) für dumm verkaufen. Zahlreichen Normen aus der 60601-Familie zwingen Hersteller dazu, immer mehr Warnungen aufzunehmen. Sind diese Warnungen denn noch an „Anwender“ gerichtet, oder nur noch an die Anwälte, die schauen wollen, ob ein Hersteller ein Versäumnis begangen hat oder eben nicht?
Bei dieser Diskussion habe ich mit keinem Wort gesagt, um welchen Wert eine Warnung denn ein Risiko senken kann. Da hilft nur ehrliches (!) und wissenschaftliches Analysieren weiter, gerne mithilfe der oben angegebenen Zahlen.
Viele Grüße
Peter Knipp
Ganz wichtige Ergänzungen durch wertvolle Hintergrundinformationen!
Herzlichen Dank, lieber Herr Knipp!
Sehr geehrter Herr Prof. Johner,
vielen Dank für Ihren interessanten und klärenden Artikel. Ich hätte hierzu eine Frage, die sich auf den Umgang der Restrisiken nach Anforderungen der MDR bezieht. Gemäß Anhang I,
– Punkt 4 „…Manufacturers shall inform users of ANY residual risks“ und auch
– Punkt 23.4 (g) „The instructions for use shall contain all of the following particulars: ANY residual risks, contra-indications…..“
wird (zumindest in der englischen Version der MDR) somit verlangt, das der Nutzer über jegliches Restrisiko informiert werden muss. Somit wäre dem Hesteller also nicht mehr möglich, entsprechende Kriterien anzusetzten, um zu entscheiden, welche Restrisiken beschrieben werden sollten und welche nicht. Bei vielen Risiken bleibt meines Wissens in der Risikoanalyse ein immer ein, wenn auch sehr geringes Restrisiko, da man nur wenige Risiken auf die Wahrscheinlichkeit „0“ („kann nach Maßnahmenergreifung nie auftauchen“) reduzieren kann.
Wie sehen Sie die Forderungen der MDR bezüglich der Beschreibung der Restrisiken (im Vergleich zur 14971)? Über eine Einschätzung Ihrerseits hierzu würde ich mich sehr freuen.
Mit freundlichen Grüßen
Kai Just
Sehr geehrter Herr Just,
danke für Ihre Frage. Ich gestehe, dass ich diese noch nicht ganz verstanden habe. Daher antworte ich etwas ins Blaue, und Sie haken einfach nach, falls ich den Punkt nicht treffen.
Dass ein Produkt auch nach risikominimierenden Maßnahmen noch Restrisiken hat, ist normal. Das ist gestattet, wenn der Hersteller zum Ergebnis kommt, dass sich diese Risiken nicht weiter reduzieren lassen und die verbliebenen Restrisiken im Vergleich zum Nutzen des Produkts beherrschbar sind. Diese Entscheidung liegt bei Ihnen.
Die verbliebenen Restrisiken müssen Sie den Anwendern kommunizieren. Da haben Sie keine Entscheidungsfreiheit.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner, erstmal vielen Dank für Ihre schnelle Antwort und entschuldigen Sie bitte, dass mich wohl etwa unverständlich ausgedrückt habe. Meine Frage zielt auf folgenden Sachverhalt.
Die 14971:2019 sagt in Kapitel 8 ….“Falls das Gesamt-Restrisiko als vertretbar beurteilt wurde, muss der Hersteller die Anwender über signifikante Restrisiken informieren, und er muss die notwendigen Informationen in die Begleitdokumentation aufnehmen, um diese Restrisiken offenzulegen.“
Und weiterhin in A2.8 (nähere Erläuterungen zu Kapitel 8): „…Der Hersteller ist dafür verantwortlich, Anwendern relevante Informationen zu signifikanten Restrisiken bereitzustellen, damit sie fundierte Entscheidungen zum Gebrauch des Medizinprodukts treffen können. Damit sind die Hersteller angewiesen, angemessene Informationen zu Restrisiken in dieBegleitdokumentation aufzunehmen. Es liegt jedoch im Ermessen des Herstellers, welche und wie viele Angaben zur Verfügung gestellt werden sollten. …..
Dies interpretiere ich so, dass der Hersteller nicht über jedes einzelne Restrisiko informieren muss, sondern – nach selbst festgelegten Kriterien – bestimmen kann, welche Restrisiken man als signifikant definiert, die man dann zur Verfügung stellen muss in der IfU, usw… Dies wird ja auch nochmal betont in dem Satz „…es liegt im Ermessen des Herstellers, welche und wie viele …“
Die MDR nach Anhang I spricht jedoch von: „ Manufacturers shall inform users of ANY residual risks“ (siehe auch mein erster Kommentar oben). Hierbei gäbe es – nach meiner Interpretation – jedoch keinerlei Spielraum, und ich müsste über jedes Restrisiko informieren. Somit müsste ich über jedes einzelne Risiko in meiner Analyse, das nach Maßnahmenergreifung nicht auf die Auftretenswahrscheinlichkeit „0“ rutscht, informieren. Denn selbst bei einer extrem geringen Wahrscheinlichkeit des Auftretens auch für sehr geringen Schaden besteht ja dann immer noch ein Restrisiko.
Sind dies nicht unterschiedliche Interpretationen, wie die Informationen über Restrisiken zur Verfügung gestellt werden müssen? Oder mache ich hier einen grundsätzlichen Denkfehler?
Mit freundlichen Grüßen
Kai Just
Lieber Herr Just,
Sie haben völlig recht. Die ISO 14971:2019 als internationale Norm formuliert die Anforderung zur Veröffentlichung der Restrisiken anders, als die MDR selbst, die in diesem Punkt strenger zu sein scheint.
Mir ist leider noch keine offizielle finale Interpretation der MDR zur Forderung „Manufacturers shall inform users of ANY residual risks“ bekannt und ich warte gespannt auf die Harmonisierung der ISO 14971 und den daraus resultierenden Z-Anhang, der hoffentlich mehr Klarheit schaffen wird.
Solange der noch fehlt, müssen wir uns also die verfügbaren Quellen an Vorgaben ansehen.
Da fällt zunächst MDCG 2019-9 ins Auge, das den SSCP beschreibt. Dort heißt es: „There is a requirement in the MDR that the IFU shall contain information on any residual risks and any undesirable side-effects, i.e. no sort of residual risk or undesirable side-effect related to the device is excluded from disclosure.“
Es liegt also die Vermutung nahe, dass man mit der MDR dem Arzneimittelbereich folgt, wo es die Verpflichtung gibt, sämtliche Risiken und Nebenwirkungen vollständig aufzulisten.
Die ISO 24971:2020 begründet sogar, warum dies notwendig ist: „The aim is to disclose information in the accompanying documentation to enable the user, and potentially the patient, to make an informed decision that weighs the residual risks against the benefits of using the medical device.“ (Anhang D.3)
Allerdings scheint die ISO 24971 auch Probleme dabei zu erkennen:
„The following are a few examples where technical practicability is questionable: […] – communicating too many residual risks so that the user has difficulty understanding which ones are really important“
Die regulatorische Seite scheint also auf dem „Manufacturers shall inform users of ANY residual risks“ zu beharren, während die Hersteller, vertreten durch Normungsgremien, eher eine sinnhafte Einschränkung der Veröffentlichung von Restrisiken fordern und das auch nachvollziehbar begründen.
Wir werden sehen, wie die Benannten Stellen sich in der Praxis hierzu bei Ihrer Konformitätsbewertung positionieren.
Der „Nationale Arbeitskreis zur Implementierung der MDR“ hat sich übrigens 2018 an einer Deutung versucht und folgendes dazu geschrieben:
(Link)
„Weiterhin bestehende akzeptable Restrisiken […] müssen dem Anwender in geeigneter Form kommuniziert werden. […] ISO 14971 gibt den Hersteller erfahrungsgemäß eine ausreichende Hilfestellung, um diese grundlegende Anforderung erfüllen zu können.“
Hier hat man damals offenbar die Diskrepanz zwischen MDR und ISO 14971 noch nicht einmal erkannt.
Fazit: Wie so oft sind die Forderungen der MDR noch nicht ausreichend in ein gemeinsames Verständnis überführt und vielleicht sind wir in 2 Jahren an dem Punkt weiter. Folgen Sie also gerne diesem Blog-Eintrag, um auf Stand zu bleiben.
Herzliche Grüße
Christian Rosenzweig
Hallo Christian,
mit der Harmonisierung der ISO 14971:2019 + A11:2021 im Dez 2021 scheint man wohl so weit zu sein wie vor der Veröffentlichung der MDR mit der „ANY residual risk“ – Anforderung.
Gemäß Annex ZA, Tabelle 1, wird die Anforderung der MDR aus „I.4, letzter Paragraph“ verlinkt mit dem zweiten Abschnitt der ISO 14971:2019, der letztendlich über die Erklärungen in TR 24971:2020 dem Hersteller die Verantwortung darüber gibt, welche Restrisiken zu veröffentlichen sind (zusätzlich zu den „significant residual risks“).
Offen bleibt nun noch, wie mit dem SSCP umzugehen ist: werden hier mehr Restrisiken aufgezeigt als in der Gebrauchsanweisung?
VG,
Jörg
Lieber Jörg,
vielen Dank für dieses wichtige und oft diskutierte Thema und Deine Frage.
Da der SSCP genauso wie die IFU quasi öffentlich zugänglich sind, sollten die dort jeweils gelisteten Restrisiken abgeglichen und konsistent sein.
Herzliche Grüße
Christian
Guten Tag Herr Prof. Johner,
vielleicht sollte man noch zusätzlich erwähnen, daß die Risikomatrix als ein Werkzeug verstanden werden sollte, wie man gezielt (ISO 13485), effizient und nicht zufällig den klinischen Stand der Technik erreichen kann.
Die MDR und MDCG fordern im CER den Beweis, daß der klinische Stand der Technik erreicht wurde.
Um das sicherzustellen, sollte der Risikograph diesen Stand der Technik berücksichtigen.
Wenn dann alle Funktionen inkl. der Schritte der Anwender, die zum Erreichen des Intended Use notwendig sind, unter Berücksichtigung dieser Grenzwerte analysiert wurden, dann hat man eine solide Grundlage für eine erfolgreiche Zulassung gelegt.
Viele Grüße,
Jörg Bigalke
Lieber Jörg, das ist tatsächlich ein wichtiger Hinweis, den wir auch in unseren Schulungen zum Risikomanagement (https://www.johner-institut.de/seminare/produktentwicklung/risikomanagement-und-iso-14971/) ausführlich behandeln. Viele Hersteller wählen eine beliebige standardisierte Risikoakzeptanzmatrix und erst durch die Klinische Bewertung am Ende des Entwicklungsprojektes zeigt sich, dass man dem Stand der Technik bzw. dem Nutzen-Risiko-Verhältnis im Vergleich zum Markt nicht gerecht geworden ist. Um den daraus entstehenden Nachteilen frühzeitig zu begegnen, sollte man die ersten Aktivitäten einer Klinischen Bewertung schon zum Projektbeginn machen und die Erkenntnisse daraus in den Risikomanagement-Plan einfließen lassen.
Herzliche Grüße
Christian Rosenzweig