
Die Risikoakzeptanzmatrix, auch Risikobewertungsmatrix genannt, ist ein bewährtes Hilfsmittel, mit dem die (Medizinprodukte-)Hersteller ihre (produktspezifischen) Kriterien für die Risikoakzeptanz ausdrücken.
Benannte Stellen prüfen häufig die Risikoakzeptanzmatrix zuerst und besonders intensiv, wenn sie die Konformität der Technischen Dokumentation mit den gesetzlichen Anforderungen bewerten.
Dieser Artikel hilft dabei, eine normen- und gesetzeskonforme Risikoakzeptanzmatrix zu erstellen und die 6 häufigsten Fehler zu vermeiden.
1. Risikoakzeptanzmatrix / Risikobewertungsmatrix – eine Übersicht
Risiken sind definiert als die Kombination aus Schweregraden von Schäden und Wahrscheinlichkeiten von Schäden. Risiken sind damit eine zweidimensionale Größe, die sich als zweidimensionale Matrix ausdrücken lassen (s. Abb. 1).
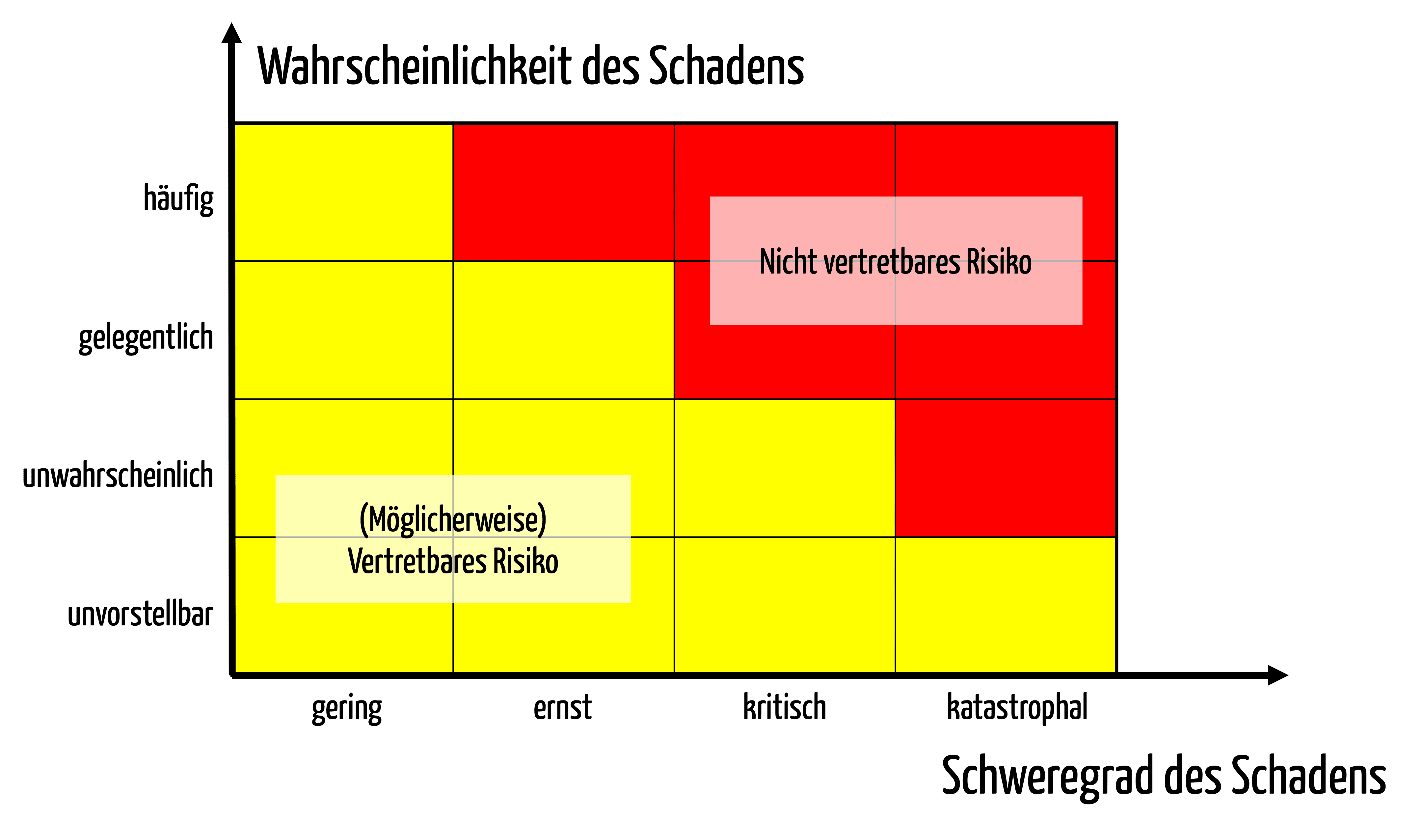
Diese Matrix teilt die Schweregrade und Wahrscheinlichkeiten in Klassen ein, typischerweise zwischen drei bis sieben Klassen für jede Dimension.
Hohe Risiken befinden sich dabei bei hohen Wahrscheinlichkeiten und hohen Schweregraden, niedrige Risiken entsprechend bei kleinen Wahrscheinlichkeiten und Schweregraden.
Die Risikoakzeptanz drücken die Hersteller durch Farbkodierung der einzelnen Zellen der Matrix aus (s. Abb. 1).
Es ist nicht konform mit der ISO 14971, die Risiken als das Produkt (die Multiplikation) von Schweregrad und Wahrscheinlichkeit zu definieren und für dieses Produkt einen Grenzwert zu bestimmen, der akzeptable und nicht-akzeptable Risiken trennt.
2. Regulatorische Anforderungen
Es gibt keine gesetzliche Anforderung, die Risikoakzeptanz in Form einer Risikoakzeptanzmatrix auszudrücken.
Auch die ISO 14971 fordert keine Risikobewertungsmatrix, auch wenn sie in älteren Ausgaben im informativen Anhang eine abbildete. In diesen älteren Ausgaben unterschied die EN ISO 14971 akzeptable Risiken, nicht akzeptable Risiken und Risiken im Bereich ALARP („as low as reasonable practical„).
Seit der 2012-er Ausgabe der EN ISO 14971 gibt es keine pauschal akzeptablen Risiken mehr. Die Unterteilung in akzeptable, nicht akzeptable und ALARP-Risiken ist somit obsolet. Vielmehr müssen Risiken „as low as reasonable possible“ sein.
Sie wecken die Aufmerksamkeit Ihres Auditors, wenn Sie noch mit einer Risikoakzeptanzmatrix arbeiten, die drei Farben (rot, gelb, grün) enthält. Denn dann ist die Wahrscheinlichkeit hoch, dass es noch generell akzeptable („grüne“) Risiken gibt. Das ist weder normen- noch gesetzeskonform.
Die FDA erwartet hingegen eine Risikoakzeptanzmatrix in der Risikomanagementakte, auch wenn sie diese Forderung nicht explizit dokumentiert hat.

Erstellen Sie eine regulatorisch konforme Risikomanagementakte – schnell und auditsicher
Mit Hilfe von duzenden Mustervorlagen und Videos zum ISO 14971-konformen Risikomanagement lernen Sie, wie Sie eine vollständige Risikomanagementakte mit allen notwendigen Dokumenten erstellen. Prüfen Sie Ihre Dokumente selbst auf Gesetzeskonformität und vermeiden Sie Fehler bei Audits und Einreichungen.
3. Tipps zum Erstellen einer Risikoakzeptanzmatrix / Risikobewertungsmatrix
a) Tipps zum Festlegen der Schweregrad-Achse
Bereits bei der Definition der Schweregradachse gibt es einige Fallen zu beachten. Zum Beispiel genügt es nicht, ein paar Bezeichnungen wie unwesentlich, leicht, schwer, kritisch und katastrophal an die Kategorien zu schreiben. Sie benötigen klare Klassifizierungsregeln, beispielsweise
- Tod (ja oder nein)
- Lebenskritischer Schaden (ja oder nein)
- Ärztliche Intervention notwendig (ja oder nein)
- Reversibler Schaden (ja oder nein)
Doch selbst damit gibt es noch offene Fragen: Was ist denn schlimmer, ein lebensbedrohlicher nicht reversibler Schaden oder ein nicht lebensbedrohlicher nicht reversibler? Ein verlorener Daumen oder ein dauerhaft behindertes Bein?
Beachten Sie unserem Fachartikel zu den Schweregrade von Schäden. Dort lernen Sie, wie Sie diese Schwergrade klassifizieren können.
Einmal mehr wird klar: Die Definition dieser ethischen Grundsätze ist keine Aufgabe der Entwicklung, sondern eine des Managements. Genau das fordert auch die ISO 14971.
b) Tipps zum Festlegen der Wahrscheinlichkeitsachse
Bei der Wahrscheinlichkeitsachse sollten Sie eine Klasse haben, welche Wahrscheinlichkeiten repräsentiert, die so gering sind, dass Ereignisse (hier Schäden) während der kompletten Lebensdauer des Produkts bei allen Produkten nicht auftreten werden. Das sind üblicherweise sehr kleine Wahrscheinlichkeiten.
Auf der anderen Seite wollen Hersteller ggf. Schäden unterschiedlich bewerten, die einmal bei jeder und einmal nur bei jeder 100. Anwendung auftreten. Dafür sind unterschiedliche Wahrscheinlichkeitsklassen die Voraussetzung.
Als Folge ergibt sich eine logarithmische Skala mit Wahrscheinlichkeitsklassen, die meist zwei Größenordnungen umfassen (s. Beispiel in Tabelle 1).
| Begriff | Beschreibung | Häufigkeit (pro Behandlung) |
| Häufig | Ein- oder mehrmals pro Behandlung | x ≥ 100 (≥ 1) |
| Wahrscheinlich | Kann bei bestimmungsgemäßem Gebrauch vorkommen | 10-2 ≤ x < 100 |
| Gelegentlich | Tritt in unregelmäßigen Abständen mehrfach pro Monat/Jahr auf | 10-4 ≤ x < 10-2 |
| Entfernt vorstellbar | Ein bis mehrmals pro Lebensdauer des Medizinprodukts | 10-6 ≤ x < 10-4 |
| Unwahrscheinlich | Nicht während Lebensdauer des Medizinprodukts | 10-8 ≤ x < 10-6 |
| Unvorstellbar | Nicht während der Lebensdauer aller Produkte. | x < 10-8 |
c) Tipps zum Festlegen der Akzeptanzkriterien
Risiken sind nur dann akzeptabel, wenn sie so gering wie möglich sind und der Nutzen die Risiken überwiegt. Genau diese Anforderung muss die Risikoakzeptanzmatrix erfüllen.
Beispiel
Angenommen es kommt auf einer Intensivstation bei jeder 10.000 manuellen Auswahl und Dosisberechnung von Medikamenten zu einem Fehler mit tödlichen Folgen z.B. aufgrund einer Kontraindikation, Medikamenten-Medikamenten-Wechselwirkung oder eines Berechnungsfehlers.
Die Zweckbestimmung einer Software bestehe darin, die Medikamentenauswahl und Dosisberechnung zu unterstützen.
Durch Fehler dieser Software oder bei der Anwendung dieser Software dürfen keinesfalls häufiger zu einer tödlichen Fehlmedikation führen. Falls der Stand der Technik nicht mehr die manuelle Auswahl und Dosisberechnung, sondern die computergestützte Medikation ist, dann müssen die Akzeptanzkriterien diesen Stand der Technik mindestens erfüllen.
Angenommen durch eine softwaregestützte Medikation tritt nur noch in jedem 100.000 Fall (10-5) eine tödliche Fehlmedikation auf, dann wären bei den Wahrscheinlichkeitskategorien gemäß Tabelle 1 maximal die Risiken in den Wahrscheinlichkeitsklassen „Unwahrscheinlich“ und „Unvorstellbar“ akzeptabel (s. Abb. 2).
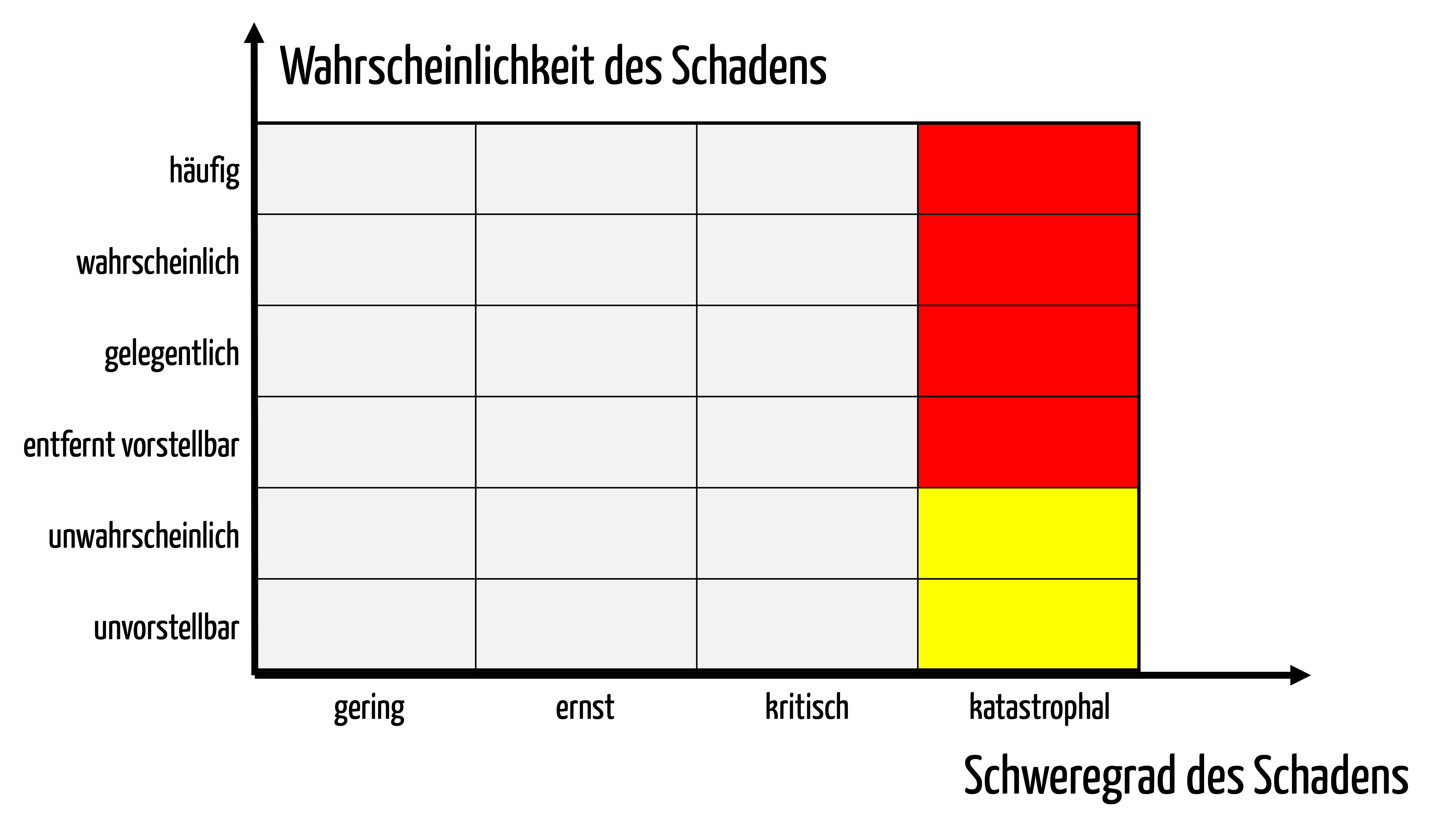
Der Hersteller muss den Stand der Technik und den Nutzen alternativer Verfahren in der klinischen Bewertung quantitativ darlegen. Damit dient die klinische Bewertung als Input für die Festlegung der Risikoakzeptanzmatrix / Risikobewertungsmatrix.
4. Häufige Fehler, die Hersteller vermeiden sollten
Fehler 1: Risikobewertungsmatrix im QM-Handbuch
Die Risiko-Nutzen-Abwägung und damit Risikobewertungsmatrix müssen produktspezifisch sein. Daher ergibt es in der Regel keinen Sinn, die Risikobewertungsmatrix im QM-Handbuch oder in einer „SOP Risikomanagement“ festzulegen.
Vielmehr sollte der Hersteller die produktspezifischen Risikoakzeptanzkriterien in einem eigenen Dokument oder im Risikomanagementplan bestimmen.
Fehler 2: Risikoakzeptanz ist nicht hergeleitet
Viele Hersteller legen die Risikopolitik eher aus dem Bauch fest. Daher ist es wichtig, den Tipp unter 3.c) zu beachten.
Fehler 3: Mit Risikoprioritätszahl arbeiten
Die ISO 14971 definiert Risiken als zweidimensionale Größe. Die Risikoprioritätszahl entspricht nicht dieser Definition. Sie darf bei FMEA angewendet werden, aber nicht zur Quantifizierung von Risiken und zur Festlegung von Risikoakzeptanzkriterien.
Fehler 4: Die Risikobewertungsmatrix nicht aktualisieren
Die Risikobewertungsmatrix muss den Stand der Technik repräsentieren (siehe Tipp in 3.c)). Der Stand der Technik ändert sich.
Beispiel: CT-Scanner
Der Nutzen von CT-Geräten orientiert sich an den verfügbaren Alternativen. In dem Maß, indem beispielsweise Kernspingeräte, die keine schädliche Strahlendosis aufweisen, immer besser in der Lage sind, Körperstrukturen zu diagnostizieren, die bisher die Domäne der CTs waren (z.B. knöcherne Strukturen), nimmt der relative Nutzen ab.
Fehler 5: Risikoakzeptanzmatrix mit „grünen“ Bereichen
Wenn die Risikomanagementakte grüne Bereiche enthält, ist das noch kein Fehler. Wenn die Farbe aber für „akzeptable“ Risiken steht, ist das nicht gesetzeskonform. Denn es gibt keine pauschal akzeptablen Risiken: Jedes Risiko muss so weit wie möglich reduziert werden.
D.h. die Farbe grün darf nur für Bereiche stehen, in denen Risiken akzeptiert werden, wenn sie maximal reduziert wurden.
Beachten Sie auch den Artikel zu den 7 häufigsten Fehlern im Risikomanagement.
Fehler 6: Wahrscheinlichkeitsachse bildet nicht den ganzen Bereich ab
Wenn Hersteller Wahrscheinlichkeitsklassen mit einer Größenordnung oder gar weniger definiert, liegt der Verdacht nahe, dass er nicht den kompletten relevanten Wahrscheinlichkeitsbereich diskutiert, wie ein Extrembeispiel zeigt:
- Häufig: p > 10-3
- Gelegentlich: 10-4 < p ≤ 10-3
- Selten: 10-5 < p ≤ 10-4
- Unvorstellbar: p ≤ 10-5
Angenommen alle Produkte eines Typs werden über deren komplette Lebensdauer 10+8-mal angewendet. Dann tritt der angeblich unvorstellbare Fall 1000 mal oder seltener auf. D.h. der Hersteller ist nicht in der Lage, zwischen Schäden z.B. mit Todesfolgen zu differenzieren, die statistisch tausendmal, die einmal oder die 0,0001 mal auftreten. Das ergibt keinen Sinn.
Er wäre zwar in der Lage, zwischen Ereignissen zu differenzieren, die mit einer Wahrscheinlichkeit von 10-4 und 10-5 auftreten. Aber so genau kann er meist gar nicht schätzen.
Grafisch ausgedrückt (siehe Abb. 3) könnte man sagen, dass sich diese Hersteller eine unnötige Differenzierungsfähigkeit bei mittleren Wahrscheinlichkeiten (analog Schweregrade) zu Lasten der „Randbereiche“ erkaufen.
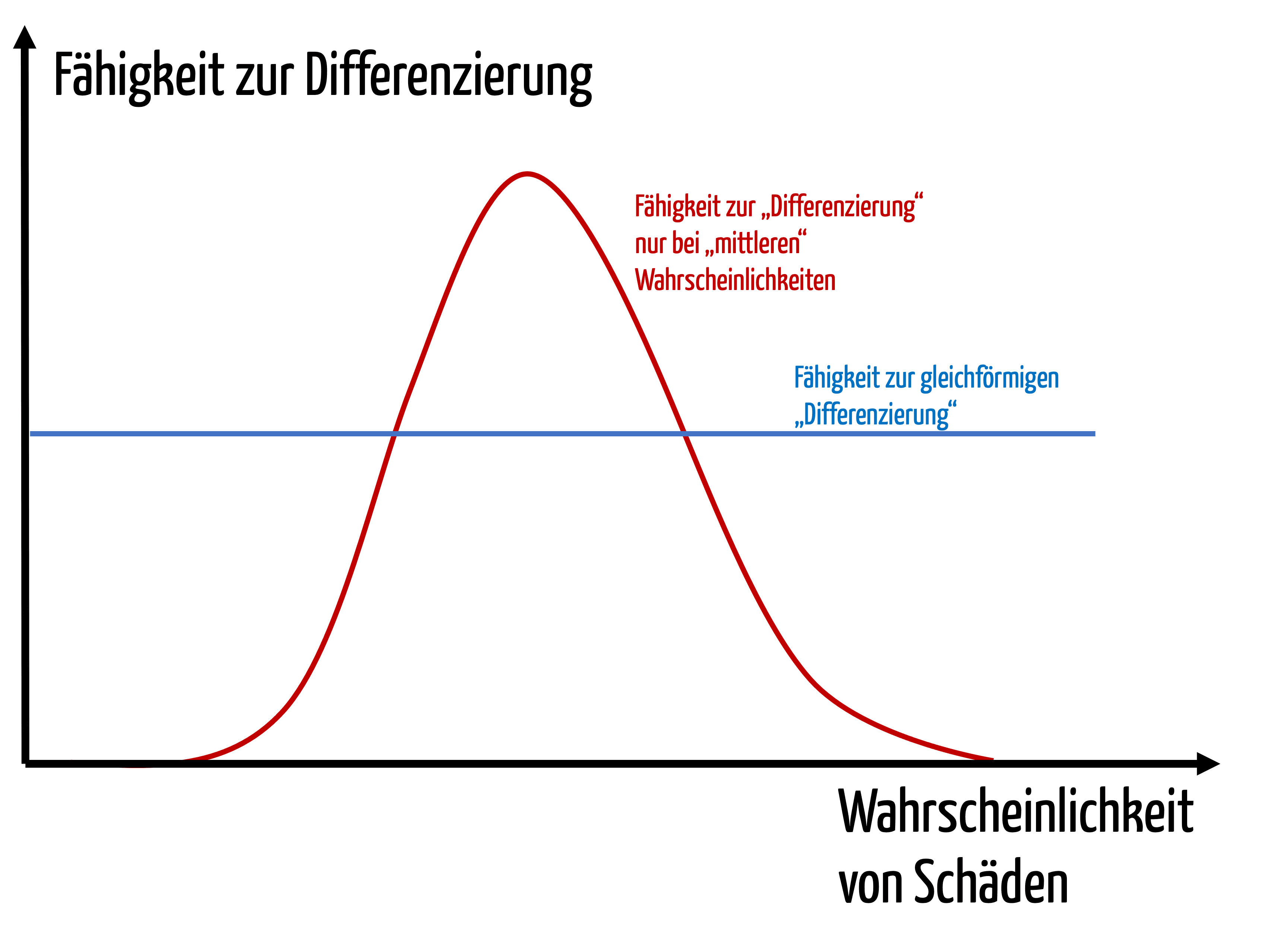
Analoge Überlegungen treffen auch auf die Schweregrad-Achse zu.
5. Fazit und Zusammenfassung
Die Risikoakzeptanzmatrix / Risikobewertungsmatrix ist ein fundamentaler Bestandteil der Risikomanagementakte. Auditoren und Prüfer erkennen oft bereits an dieser Matrix, ob die Hersteller die Konzepte des Risikomanagements und des Nutzen-Risikoverhältnisses verstanden haben.
Die Risikoakzeptanzmatrix drückt dieses Nutzen-Risikoverhältnis spezifisch für das jeweilige Produkt aus. Sie muss auch in Übereinstimmung mit der Risikopolitk und den Ergebnissen der klinischen Bewertung stehen.
Weil die Risikobewertungsmatrix auch den Stand der Technik widerspiegelt, müssen sie die Hersteller regelmäßig (mindestens jährlich) auf Gültigkeit prüfen.
Unterstützung!
Das Johner Institut unterstützt Hersteller nicht nur beim Erstellen der Risikoakzeptanzmatrix, sondern bei der ganzen Risikomanagementakte.
Das Seminar „Risikomanagement und ISO 14971“ verschafft einen schnellen Einstieg in das Thema. Dabei lernen die Teilnehmenden auch Risikoakzeptanzmatrizen zu erstellen und zu bewerten.

Sie schreiben unter „Fehler 5″: Risikoakzeptanzmatrix mit „grünen“ Bereichen“, Jedes Risiko muss so weit wie möglich reduziert werden. In der 14971:2019 steht aber, dass wenn das evaluierte Risiko bereits akzeptable ist, keine der Anforderungen aus den Kapiteln 7.1 – 7.5 angewandt werden muss. Steht das nicht im Widerspruch zu Ihrer Aussage?
6 Risk evaluation
For each identified hazardous situation, the manufacturer shall evaluate the estimated risks and
determine if the risk is acceptable or not, using the criteria for risk acceptability defined in the risk
management plan.
If the risk is acceptable, it is not required to apply the requirements given in 7.1 to 7.5 to this hazardous
situation (i.e., proceed to 7.6) and the estimated risk shall be treated as residual risk.
Sehr geehrter Herr Eichhorn,
danke für die spannende Frage!
Regulatorisch relevant ist nicht (nur) die ISO 14971, sondern die EN ISO 14971 und deren Z-Anhänge. Diese Z-Anhänge diskutieren, wie vollständig die entsprechenden gesetzlichen Anforderungen (hier der MDR) erfüllt sind.
Die MDR fordert beispielsweise:
Genau dieser Punkt, den Sie mit Recht diskutieren, war Ausgang von Auseinandersetzungen zwischen Kommission und Normenkommittee, die zur „fast über Nacht erschienenen“ EN ISO 14971:2012 führten. Diese Norm hatte dann im Anhang die Interpretation, auf die Sie im Artikel gestoßen sind.
Beste Grüße, Christian Johner
Sehr geehrte Damen und Herren,
dann scheint der gesamte Medizintechniksektor das Risikomanagement nach ISO 14971 aber wirklich nicht richtig zu machen, wie ich auch immer sage, denn:
I) die Felder (Kombinationen) der Risiko-Akzeptanzmatrix ist nicht wissenschaftlich und fakten-basiert hergeleitet
II) eine quantitative Methode wird nicht verwendet und hierzu muss man mit Wahrscheinlichkeiten multiplizieren
III) die Restrisiken werden nicht bestimmt (kein einziger Hersteller summiert die einzelnen Restrisiken zusammen
IV) die Risikopolitik ist zu generell formuliert um die Akzeptanzmatrix-Kriterien hierdurch herzuleiten
V) Keine Verifikation und Validierung, dass Restrisiken soweit nur irgend möglich reduziert wurden
Kurzum, jedes Risikomanagement aller Medizintechnikfirmen wurde und wird also faktisch nicht gemacht nach MDD und MDR, denn die Risikomatrix ist das Herz und Kernstück des gesamten Risikomanagements. Wenn diese nicht stimmt, dann ist das Risikomanagement quasi gar nicht richtig gemacht worden – also wirklich NICHT gemacht worden.
Somit dürfe kein einziges Medizintechnikprodukte in Verkehr gebracht werden (= verkauft werden in EU), was man auch nicht umsetzen kann, weil man diese braucht, auch wenn das Risiko nicht richtig minimiert wurde in jedem Fall.
Dieses Dilemma kann nur die Politik lösen indem Sie die RA QM QA UND RM Dokumentationspflichten verstärkt, unterstütz, erhöht und nicht abbaut.
Qualitätsmanager und Risikomanager und Regulatory Affairs müssen mehr im Unternehmen zu sagen haben um diese Verpflichtungen umsetzen zu können.
Die Lean und Business Leute sowie McKinsey, PwC, alle BWLer, und Strategieberater im Hintergrund verhindert, dass die Produkte sicher sind mit gutem Risikomanagement nach ISO 14971 uns setze alle maximal unter Druck das nicht richtig zu machen!
Business ist der Feind von Qualität und Sicherheit im Sektor und daher machen es alle falsch, und werden gezwungen es falsch zumachen.
Mit freundlichen Grüßen,
Vielen Dank für Ihren Kommentar zum derzeitigen Stand der regulatorischen Compliance. Tatsächlich werden die Anforderungen teilweise nicht korrekt umgesetzt. Das mag manchmal daran liegen, dass die Hersteller die ISO 14971 nur als Vorlage nehmen, den Prozess aber individuell auslegen oder anpassen. Das dürfen sie auch, denn es handelt sich nur um eine harmonisierte Norm, kein Gesetz. Eigentlich müssten sie dann aber gegenüber den Behörden und Benannten Stellen eine schriftliche Begründung liefern, warum sie glauben, das zugrundeliegende Gesetz mit ihrem individuellen Vorgehen zu erfüllen. Das fordern bisher nicht alle Benannten Stellen.
Und da haben wir das nächste Problem: Medizinprodukte der Klasse I benötigen noch nicht mal eine Konformitätsprüfung durch eine Benannte Stelle. Eine Großzahl an Risikomanagementakten wurde also noch nie durch externe Instanzen auf Konformität geprüft!
Doch ich möchte diese Aussage auch gleich wieder relativieren: Wir erleben in unserer Beratungspraxis ständig und regelmäßig Hersteller, die von den lokalen Behörden oder eben doch von der Benannten Stelle auf Fehler in der Risikomanagementakte aufmerksam gemacht werden. Und zwar unter anderem genau die Punkte, die Sie in Ihren Ausführungen aufzählen.
Ich würde also aus meiner Perspektive festhalten:
– Einige Risikomanagementakten im Markt haben tatsächlich noch nicht den geforderten Reifegrad an regulatorischer Konformität.
– Es ist nur eine Frage der Zeit, bis das Behörden oder Benannten Stellen auffällt und sie entsprechende Korrekturen einklagen. Immer häufiger sogar mit Androhung von Zertifikatsentzug oder Vermarktungsverbot.
– Wir haben im Bereich Risikomanagement keinen Handlungsbedarf, die regulatorischen Anforderungen anzupassen, gar zu erhöhen. Wir haben eher die Notwendigkeit, die Umsetzung strenger und systematischer zu überwachen.
Ich sehe das Thema daher nicht so pessimistisch und freue mich, dass in meinen Seminaren zum Risikomanagement immer wieder wissbegierige und motivierte Menschen sitzen, die die richtige Vorgehensweise erlernen möchten.