
Die Frage, ob eine Risikominimierung durch Informationen erlaubt ist, führt regelmäßig zu Diskussionen. Die Antwort auf diese Frage ist wichtig, denn sie entscheidet über Konformität und Nicht-Konformität der Medizinprodukte.
Dieser Artikel gibt die Antwort und löst damit ein „historisches Missverständnis“ auf.
1. Regulatorischer Rahmen
Alle Hersteller sind verpflichtet, die Risiken, die durch ihre Medizinprodukte entstehen, zu minimieren. Bezogen auf den Nutzen des Produkts müssen die Restrisiken akzeptabel sein.
Um die Risiken zu minimieren, gibt es mehrere Typen an Maßnahmen. Gesetze wie MDR und IVDR sowie die Risikomanagementnorm ISO 14971 nennen diese Typen und die Reihenfolge, in denen sie zur Anwendung kommen müssen (s. Abb. 1).
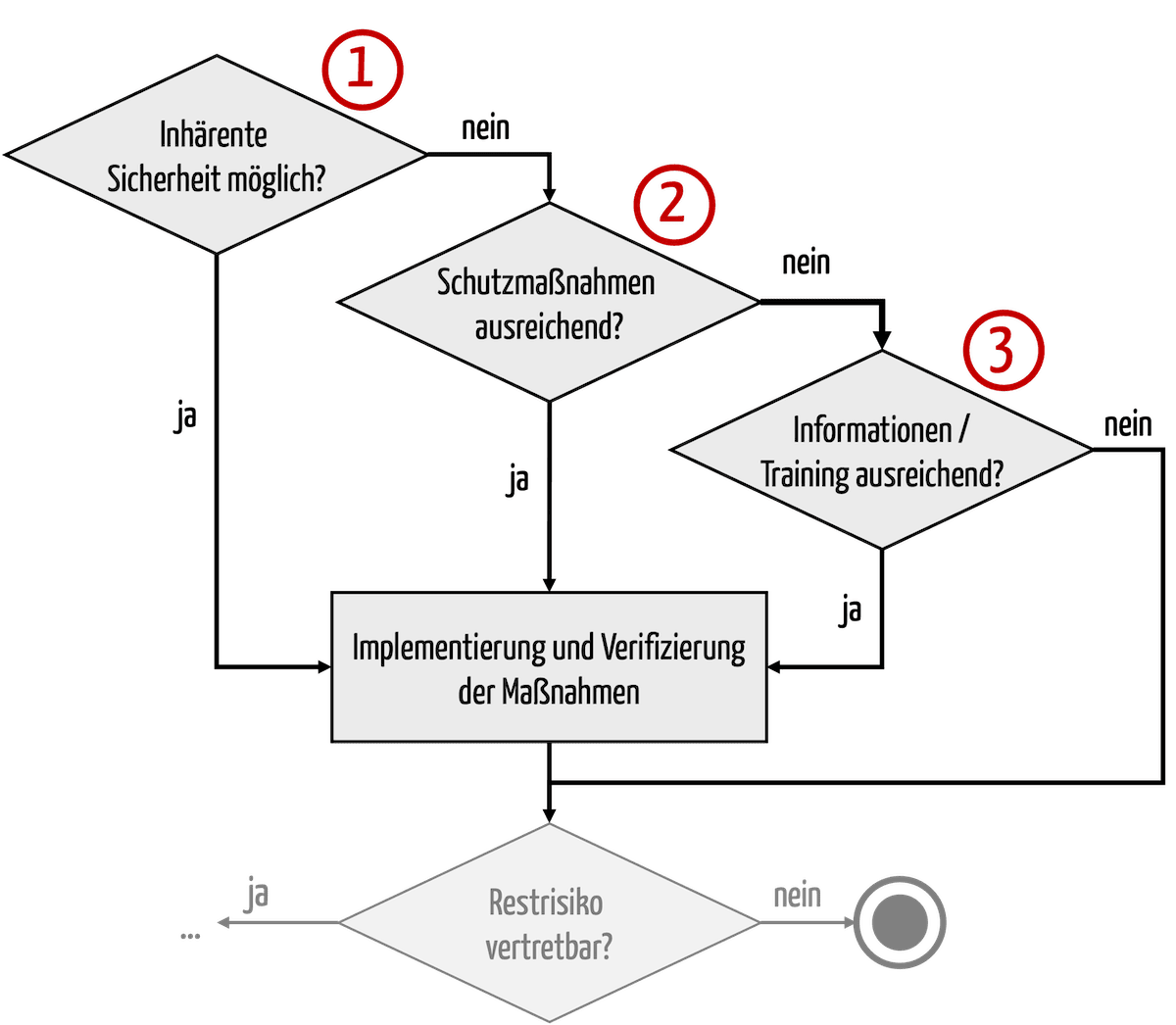
Es gilt somit kein generelles Verbot einer Risikobeherrschung bzw. Risikominimierung durch Informationen.

Erstellen Sie eine regulatorisch konforme Risikomanagementakte – schnell und auditsicher
Mithilfe von Mustervorlagen und Videos lernen Sie, wie Sie eine vollständige Risikomanagementakte mit allen notwendigen Dokumenten erstellen. Prüfen Sie Ihre Dokumente selbst auf Gesetzeskonformität und vermeiden Sie Fehler bei Audits und Einreichungen.
2. Typen an Informationen
Um die Frage zu beantworten, ob eine Risikominimierung durch Informationen erlaubt ist, hilft eine Übersicht über die verschiedenen Typen:
- Warnungen am Produkt, z. B. Aufkleber oder Pop-ups bei Software-Oberflächen
- Hinweise in Gebrauchsanweisungen und anderen Begleitmaterialien zur richtigen Nutzung des Produkts
- Informationen in Gebrauchsanweisungen über Restrisiken
- Schulungen von Anwendern
3. Auslöser der Verwirrung
Bei der Harmonisierung der ISO 14971:2012 schrieben die Anwälte des Harmonisierungsdienstleisters in den Anhang Z:
… manufacturers shall not attribute additional risk reduction to the information given to the users…
Anhang Z der ISO 14971
Dabei haben die Anwälte leider zwei Typen von Informationen in einen Topf geworfen:
- Informationen, die „nur“ alle Restrisiken auflisten. Dies ist eine Forderung der MDR in Anhang I, Abschnitt 4 („Manufacturers shall inform users of any residual risks.“).
- Informationen, die konkrete und spezifische Maßnahmen zur Minimierung von Risiken darstellen. Diese enthalten konkrete Handlungsvorgaben für Personen im Umgang mit dem Produkt.
4. Die Auflösung
Die Liste der Restrisiken ist …
- vergleichbar dem Beipackzettel mit den Nebenwirkungen bei den Medikamenten,
- regulatorisch vorgeschrieben und
- keine (!) risikominimierende Maßnahme.
Handlungsanweisungen für die Anwender …
- sind regulatorisch vorgeschrieben, wenn damit Risiken minimiert werden können,
- zählen dann zu den risikominimierenden Maßnahmen und
- sind als solche auch erlaubt.
Auch das Team-NB teilte in einem Consensus Paper bereits 2014 diese Einschätzung.
Bei jedem elektrisch betriebenen Medizinprodukt besteht das Risiko eines elektrischen Schlags. Dieses (Rest-)Risiko besteht auch, wenn der Hersteller alle von der IEC 60601-1 vorgeschriebenen Maßnahmen (z. B. zu Luft- und Kriechstrecken) erfüllt. Dieses Risiko muss der Hersteller in die Liste der Restrisiken eintragen und veröffentlichen.
Der Anwender kann diese Information bei seiner risikobasierten Entscheidung für oder gegen die Verwendung des Produktes berücksichtigen. Den Stromschlag selbst (z. B. bei einem Produktfehler) kann er nicht verhindern. Demnach ist es auch keine risikominimierende Maßnahme, diese Information an den Anwender zu geben.
Wenn der Hersteller hingegen in der Gebrauchsanweisung vorschreibt, dass der Techniker vor der Reparatur und vor dem Öffnen des Gehäuses den Netzstecker ziehen muss, dann ist das eine klare Handlungsanweisung, die bei Erfüllung das Risiko minimiert. Nämlich das Risiko, bei Kontakt mit elektrischen Bauteilen im Inneren des Produkts einen Stromschlag zu bekommen.
5. Fazit
Risikominimierung durch Informationen ist also durchaus erlaubt. Zu unterscheiden sind:
- Informationen, die alle Restrisiken auflisten
Die Veröffentlichung der Liste der Restrisiken ist regulatorisch vorgeschrieben und keine risikominimierende Maßnahme. - Handlungsanweisungen für die Anwender
Sie sind regulatorisch vorgeschrieben, wenn damit Risiken minimiert werden können. Als risikominimierende Maßnahme sind sie erlaubt.
Die Umsetzung und die Wirksamkeit von risikominimierenden Maßnahmen müssen die Hersteller nachweisen. Risikominimierende Maßnahmen durch Informationen werden i. d. R. durch Usability-Tests überprüft.
Trainingsunterlagen und Gebrauchsanweisungen zählen zum User Interface und sind daher Gegenstand der regulatorischen Anforderungen an die Gebrauchstauglichkeit und im Anwendungsbereich der IEC 62366-1.
Haben Sie noch Fragen zum Risikomanagement? Dann nutzen Sie
- das kostenlose Micro-Consulting des Johner Instituts oder
- die Hilfe des Risikomanagement-Teams beim Erstellen und Prüfen Ihrer Risikomanagementakte.
So stellen Sie sicher, dass es keine Probleme bei Audits und Prüfungen Ihrer Technischen Dokumentation gibt und keine Verzögerungen bei der Zulassung Ihrer Produkte auftreten.
Lesen Sie hier mehr zur Risikominimierung und hier mehr zum Risikomanagement im Allgemeinen.

Lieber Herr Prof. Johner,
vielen Dank für die Darstellung dieses „historischen Missverständnisses“, das sich mit der aktuellen Ausgabe der EN ISO 14971 ja auch ein Stück weit aufgelöst hat.
In der Praxis hatte diese Fragestellung aus meiner Sicht nur begrenzte Relevanz – zumindest bei den Medizinprodukten in meinem Umfeld. Dass eine Gebrauchsanweisung für viele Produkte oft nicht (mehr) gelesen wird, ist sicher nicht abzustreiten.
Außerdem ist es oft nicht einfach, die Wirksamkeit nachzuweisen. Nur weil in einer Gebrauchstauglichkeitsstudie mit 15 Teilnehmern kein Teilnehmer einen Fehler gemacht hat, ist die Wirksamkeit einer Maßnahme zur Reduzierung eines Risikos von „kommt 10 Mal in 1 Mio. Fällen vor“ auf „kommt weniger als 1 Mal in 1 Mio. Fällen vor“ noch nicht erbracht.
In der Praxis kann es sich daher lohnen, den Handlungsanweisungen nicht unbedingt eine Risikominimierung zuzuschreiben, zumindest nicht, wenn es die einzige mögliche Maßnahme ist.
Auf der anderen Seite kann ich auch erkennen, dass es einige Produkte gibt, bei denen es durchaus sinnvoll sein kann. Das Wartungshandbuch für den Servicetechniker ist ein gutes Beispiel für Informationen, die sicherlich ausführlicher gelesen und durchgearbeitet werden.
Wie immer, vielen Dank für den Beitrag!
Viele Grüße!
Torsten Kneuss
Lieber Herr Kneuss,
vielen Dank für Ihre wertschätzenden und wichtigen Anmerkungen!
Herzliche Grüße
Christian Rosenzweig
Sehr geehrter Herr Rosenzweig,
Ich habe ein paar Kommentare zu Ihrem Text:
– In Kapitel 3 vergleichen Sie die EN ISO 14971:2012 mit der MDR. Das können Sie in diesem Beispiel nicht machen, denn der Z-Annex der EN ISO 14971:2012 bezog sich auf die MDD
– Egal was die Sinnhaftigkeit des Verbotes der Benutzung von Handlungsanweisungen als risikomindernde Massnahmen durch den Z-Annex betrifft; Die NBs haben sich unter der MDD darauf berufen und solche Massnahmen in meinem Umfeld nicht akzeptiert.
– Die Informationen über die Restrisiken war in der MDD (und aktuell auch in der MDR) gefordert, aber nicht als risikomindernde Massnahme gelistet und können auch nicht als solche benutzt werden (auch wenn sie zumindest teilweise so gewirkt haben).
– Seit der MDR und der EN ISO 14971:2019 ist der damalige Z-Annex nicht mehr vorhanden.
Mit der MDR wurde dieses „Verbot“ wieder teilweise aufgehoben (MDR, GSPR 4 c)). Jetzt dürfen Sicherheitsinformationen wieder als risikomindernde Massnahmen benutzt werden, aber nicht als reine Handlungsanweisungen sondern nur als „Warnungen, Vorsichtshinweise, Kontraindikationen“. Da diese Aufzählung in einer Klammer steht, ohne dass sie als Beispiele bezeichnet werden (wie in anderen Klammern in der MDR), muss davon ausgegangen werden, dass diese die einzigen Möglichkeiten darstellen, wie Sicherheitsinformationen als risikomindernde Massnahmen benutzt werden können.
Wenn das Ganze aber realistisch angeschaut wird, sind eigentlich nur prominent am Medizinprodukt platzierte Warninformationen und eine intensive Schulung in der Realität einigermassen funktionierende risikomindernde Massnahmen. Alles, was in der Gebrauchsanweisung steht erfüllt zwar die Regularien und Normen, aber ich bin der Meinung, dass diese einerseits meistens nicht gelesen wird und andererseits auch bei der Usability-Validierung durch die begrenzte Anzahl Teilnehmer nicht wirklich geprüft wird.
Viele Grüsse aus der Schweiz
Andreas Meschberger
Sehr geehrter Herr Meschberger,
besten Dank für Ihre wichtigen Anmerkungen! Da Christian Rosenzweig im Urlaub ist, antworte ich an seiner statt.
Sie haben völlig Recht mit Ihrer Aussage im ersten und dritten Spiegelstrich, dass die Z-Anhänge sich auf die MDD und nicht MDR bezogen. Die Aussage in dem von Ihnen referenzierten 3. Kapitel ist, dass Anwälte(!) diese Parallelen gezogen haben, nicht wir.
Ihrer Aussage in Ihrem zweiten Spielstrich stimmen wir ebenfalls zu. Das ist genau das was der Artikel zum Ausdruck bringen sollte.
Der einzige Punkt, bei dem ich noch nicht sicher bin, ob wir das gleiche Verständnis haben, ist Ihre Aussage mit dem Aufheben des Verbots. Ein Verbot, sicherheitsbezogene Hinweise als risikominimierende Maßnahmen zu nutzen, hat es in dieser Form nie gegeben. Dieses Missverständnis entstand durch eine etwas unglückliche Formulierung und Auslegung viele Benannter Stellen.
Ich teile völlig Ihre Einschätzung, dass — unabhängig von der Möglichkeit, sicherheitsbezogen Informationen als risikominimierende Maßnahmen zu nutzen — die Wirksamkeit genau dieser Maßnahmen beschränkt ist (z.B. weil sie nicht gelesen werden) und unbedingt nachgewiesen werden müssen (z.B. durch Usability Tests).
Nochmals vielen Dank für Ihre wertvollen Gedanken!
Viele Grüße
Christian Johner
Hallo an das Johner Team,
eine Ergänzung zu Informationen auf einer GUI:
Bezüglich Eures Kapitel 2 „Typen an Informationen“ lohnt auch noch ein Blick in die IEC 62366-1, Table B.2. Dort wird eine software message incl. confirmation step als protective measure verstanden. Möglicherweise also eine sinnvollere Risikominimierung im Vergleich zur reinen Information in Form einer Warnung per Popup.
Viele Grüße
Krumholz
Vielen Dank für diese Ergänzung, Herr Krumholz!