
Ohne eine gesetzeskonforme Leistungsbewertung von In-vitro-Diagnostika (IVD) riskieren Hersteller nicht nur Probleme bei der Zulassung; sie riskieren die Sicherheit der Patienten. Daher stellen Regularien wie die IVDR hohe Anforderungen an die Leistungsbewertung (performance evaluation).
Erfahren Sie, welche Anforderungen genau die IVDR an die Leistungsbewertung stellt, um die Konformität Ihres Produkts zu erreichen, und wie sich diese von den Vorgaben der IVDD unterscheiden.
In acht Schritten lassen sich die häufigsten Fehler vermeiden.
1. Weshalb die Leistungsbewertung von IVD so wichtig ist
In diesem Kapitel erfahren Sie, welche negativen Folgen eine schlechte Leistungsbewertung haben kann. Diese sollten Sie als IVD-Hersteller unbedingt vermeiden.
a) Die Aufgabe von In-vitro-Diagnostika
In-vitro-Diagnostika (IVD) haben das Ziel, aus menschlichen Proben wie Blut oder Gewebe Informationen zu liefern, die Rückschlüsse auf beispielsweise physiologische oder pathologische Prozesse im Körper zulassen.
Ein IVD soll z. B. bestimmen:
- Tumormarker im Blut
- Coronavirus (SARS-CoV-2) in einem Abstrich
- Krebszellen in einer Biopsie
Diese Informationen sind entscheidend für die Diagnose und damit für die weitere Therapie eines Patienten. Die Leistungsbewertung von IVD muss sicherstellen, dass die bereitgestellten Informationen korrekt, präzise und genau sind und den beabsichtigten klinischen Nutzen erbringen.
b) Mögliche Fehler von IVD
Die Informationen, die durch Anwendung eines IVDs erzeugt werden, können jedoch in mehrfacher Hinsicht fehlerhaft sein:
- Sie sind falsch.
- Falsch-positiv: Ein Labortest sagt fälschlicherweise, dass ein Untersuchungsergebnis auffällig oder krankhaft ist, obwohl keine Krankheit vorliegt.
- Falsch-negativ: Ein Labortest sagt fälschlicherweise, dass ein Untersuchungsergebnis nicht auffällig oder krankhaft ist, obwohl eine Krankheit vorliegt.
- Die Informationen sind ungenau.
- Das IVD zeigt die Informationen gar nicht oder verspätet an.
Die Ursachen für solche Fehler sind mannigfaltig. Beispiele sind:
- Das Analyseverfahren ist generell ungeeignet.
- Der Hersteller hat nicht alle Einflussfaktoren in den Gewebeproben beachtet, z. B. den Einfluss von Medikamenten.
- Das für die Entwicklung und die Leistungsstudien verwendete Probenmaterial entspricht nicht der tatsächlichen Patientenpopulation.
- Reagenzien verändern sich, z. B. unter dem Einfluss von Wärme, Sauerstoff oder Licht.
- Software-Fehler führen zum Vertauschen von Patienten.
- Beim Öffnen der Proben führen kleinste Spritzer zu einer Kreuzkontamination anderer Proben.
c) Folgen für die Patienten
Es besteht die Gefahr, dass fehlerhafte Informationen weitreichende Folgen für Patienten haben.
Falsch-positive Ergebnisse lösen meist eine unnötige Kaskade von Maßnahmen aus, die den Patienten Schaden oder zumindest unnötigen Stress (Angst) zufügen:
- Unnötige Blutentnahmen
- Vermeidbare Biopsien
- Falsche Therapien, z. B. mit Medikamenten oder gar Operationen
Falsch-negative Ergebnisse hingegen führen zu kritischen Verzögerungen dringend erforderlicher Therapien oder haben gar Einfluss auf die öffentliche Gesundheit.
d) Folgen für die Hersteller
Mit der Leistungsbewertung stellen die IVD-Hersteller nicht nur sicher, dass die Risiken für die Patienten minimiert sind, sondern auch die Folgen für das eigene Unternehmen.
- Das Produkt „fällt bei der Zulassung durch“ und kommt dadurch später auf dem Markt. Dadurch gehen dem Unternehmen geplante Umsätze verloren.
- Das Produkt muss vom Markt genommen werden. Das hat nicht nur finanzielle Auswirkungen, sondern schadet der Reputation. Solche Rückrufe müssen die Hersteller auf den Webseiten der Behörden veröffentlichen.
- Auf die Hersteller kommen im schlimmsten Fall Schadensersatzforderungen geschädigter Patienten zu.
- Neben unzufriedenen Kunden und einem beschädigten Image sollten die Hersteller auch die zusätzlichen Kosten für den Support und Nachbesserungen nicht vergessen.
2. Was eine Leistungsbewertung ist und was sie nachweisen muss
In diesem Kapitel lernen Sie wichtige Konzepte und Definitionen kennen, die Sie benötigen, um die Gesetzestexte zu verstehen.
a) Definitionen
Die IVDR weist zum Glück zahlreiche Definitionen auf:
Diese Definition verwendet weitere definierte Begriffe:
- Wissenschaftliche Validität
- Analyseleistung
- Klinische Leistung
Die Basis für jedes IVD stellt der Analyt dar, anhand dessen ein physiologischer Zustand bzw. ein Krankheitsbild erkannt werden soll. Beispielsweise besteht ein Zusammenhang zwischen dem Coronavirus (SARS-CoV-2) und der Krankheit Covid-19. Ebenso gibt es einen Zusammenhang zwischen dem im Blut gemessenen Gehalt an Prostata-spezifischem Antigen (PSA) und dem Risiko eines Mannes, an Prostatakarzinom erkrankt zu sein.
Um sicherzustellen, dass der Analyt auch zuverlässig mit der gewählten Methode gemessen werden kann, prüfen die IVD-Hersteller die analytische Leistung des Produkts. Um die Beispiele fortzuführen: Die Analyseleistung eines IVDs liegt darin, das Coronavirus möglichst reproduzierbar und in geringen Konzentrationen korrekt zu identifizieren oder die PSA-Konzentration möglichst präzise zu bestimmen.
Die Leistung eines IVDs erfordert neben der Analyseleistung auch den Nachweis der „klinischen Leistung“. Das ist ein weiterer Begriff, der zum Verständnis notwendig ist.
b) Klinischer Nachweis
Den Nachweis des klinischen Nutzens eines IVDs müssen IVD-Hersteller auf Grundlage von Daten zur wissenschaftlichen Validität, zur Analyseleistung und zur klinischen Leistung erbringen. Die wissenschaftliche Validität betrachtet die Assoziation von Analyt und Krankheitsbild bzw. physiologischem Zustand. Eine zuverlässige und präzise Methode zur Messung des Analyten ist schließlich Voraussetzung, dass das IVD die klinische Leistung erbringt. Sie belegt die Fähigkeit des IVDs, klinische, physiologische oder pathologische Zustände bzw. Vorgänge zu bestimmen, und stellt damit einen wesentlichen Aspekt der Leistung des Produkts dar.
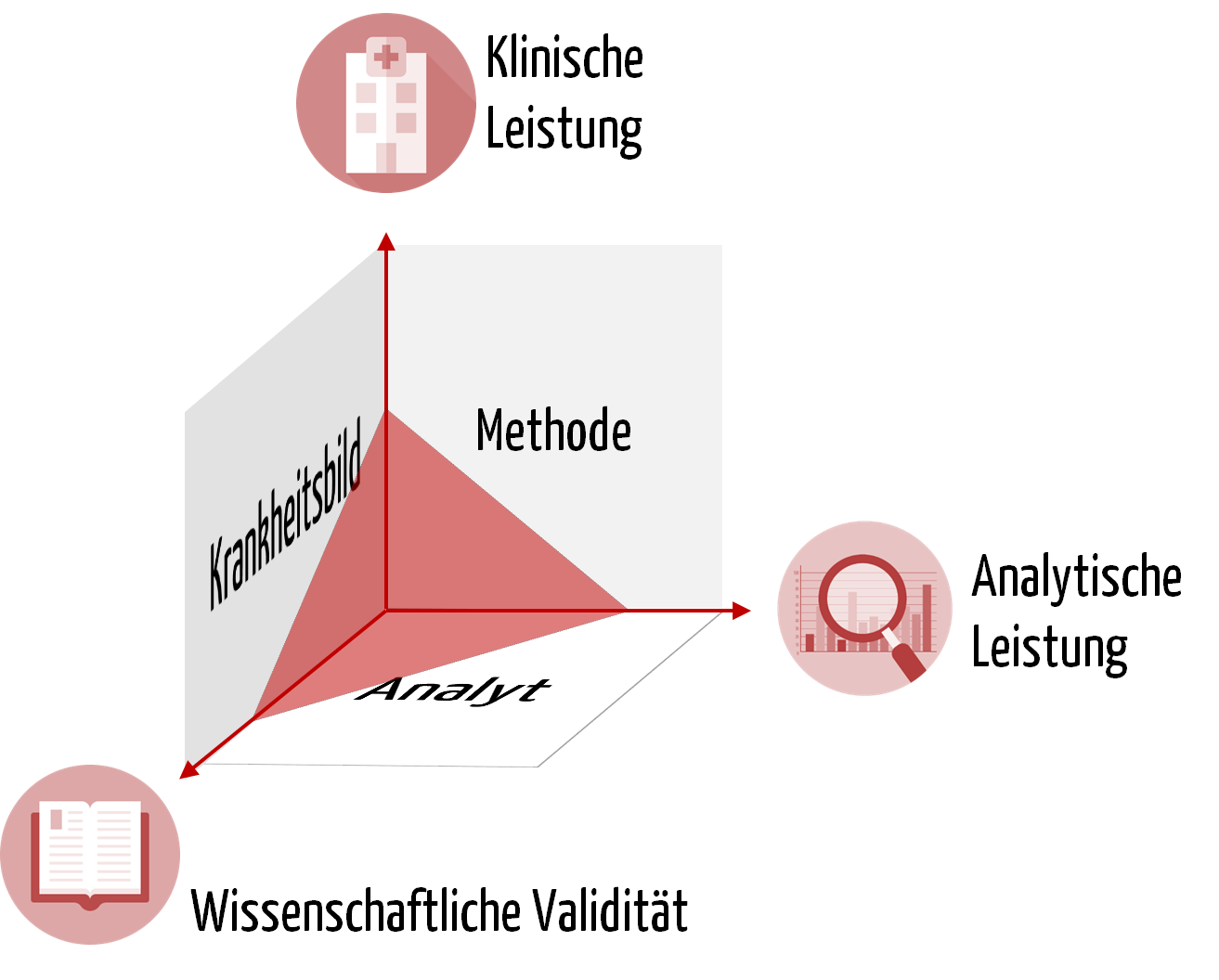
Wichtig ist zudem die Feststellung, dass die klinische Leistung keine absolute Fähigkeit ist, sondern von der Zweckbestimmung und damit von der „bestimmten Zielbevölkerung“ und den „bestimmten vorgesehenen Anwendern“ abhängt.
Die klinische Leistung eines IVDs mag beispielsweise für „normale“ Patienten gut sein, aber nicht für Patienten, die sich gerade einer Chemotherapie unterziehen, weil die Zytostatika die Messgenauigkeit beeinflussen.
Die Leistung eines Produkts mag für professionelle Anwender hervorragend sein, für Laienanwender aber nicht. Daher betont die IVDR, dass die Leistung eines Produkts von der Zweckbestimmung abhängig ist.
Der klinische Nachweis, also der Beweis, dass ein Produkt sicher ist und den beabsichtigten klinischen Nutzen erzielt, wird durch die Leistungsbewertung erbracht. Diese Leistungsbewertung bewertet die klinische Leistung, die Analyseleistung und die wissenschaftliche Validität.
3. Welche regulatorischen Anforderungen an die Leistungsbewertung IVD-Hersteller beachten müssen
In diesem Kapitel erhalten Sie einen Überblick über
- die relevanten Regularien (ohne diese erst recherchieren zu müssen) und
- die wichtigsten Anforderungen.
Sie erfahren auch, welche Unterschiede es zwischen der IVDD und IVDR gibt und wie Sie diesen begegnen können.
Wie bereits die IVD-Richtlinie 98/79/EG (IVDD), so verpflichtet auch die IVD-Verordnung 2017/746 (IVDR) im Anhang I dazu, die grundlegenden Sicherheits- und Leistungsanforderungen einzuhalten. Zu deren Nachweis zählt die Leistungsbewertung.
a) IVDR
Artikel 5
Diese Aussage macht die IVDR wörtlich:
Um ein IVD in Verkehr zu bringen oder in Betrieb zu nehmen, müssen die für das Produkt relevanten grundlegenden Sicherheits- und Leistungsanforderungen erfüllt werden. Zum Nachweis der Erfüllung der Anforderungen muss u. a. eine Leistungsbewertung durchgeführt werden.
Artikel 5, IVDR
Artikel 56
Der Artikel 56 fordert, dass die Hersteller prüfen, ob die grundlegenden Sicherheits- und Leistungsanforderungen gemäß Anhang I erfüllt sind. Zu dieser Prüfung müssen die Hersteller auch eine Leistungsbewertung durchführen.
Diese Leistungsbewertung umfasst (wie oben bereits dargestellt) den Nachweis der wissenschaftlichen Validität des Analyten, der Analyseleistung und der klinischen Leistung.
Dabei stehen im Vordergrund insbesondere die Nachweise
- der allgemeinen Anforderungen gemäß Anhang I Kapitel I,
- der Leistungsmerkmale in Anhang I, Abschnitt 9,
- eines akzeptablen Nutzen-Risiko-Verhältnisses sowie
- der Kombination mit anderen Produkten oder Ausrüstungen.
Der Artikel verweist für die Leistungsbewertung auf die Vorgaben in Anhang XIII, Teil A.
Anhang XIII und ISO 20916:2019
Dieser Anhang legt fest, wie IVD-Hersteller die Leistungsbewertung planen, durchführen und dokumentieren müssen. Dazu sollten die Hersteller auch die ISO 20916:2019 beachten, die die berichtigte IVDR vom 03.05.2019 direkt referenziert.
„Die Bestimmungen über Leistungsstudien sollten den fest etablierten internationalen Leitlinien in diesem Bereich entsprechen, wie der in Entwicklung befindlichen internationalen Norm ISO 20916 über klinische Leistungsstudien, bei denen Proben von Menschen verwendet werden, damit …“
IVDR, Erwägungsgrund 66
Die ISO 20916:2019 wurde im Mai 2019 veröffentlicht. Sie trägt den Titel „In vitro diagnostic medical devices – Clinical performance studies using specimens from human subjects – Good study practice“.
Die ISO 20916:2019 beschreibt im Detail die Anforderungen an die Planung und Durchführung von klinischen Leistungsstudien. Sie ergänzt die IVDR in der Beschreibung der Organisation, der beteiligten Rollen und den Vorgaben zur Durchführung einer solchen Studie.
Überlappungen mit der IVDR gibt es insbesondere in Bezug auf die Inhalte des klinischen Leistungsstudienplans (gemäß IVDR, Anhang XIII, Abschnitt 2.3.2.). Diesen Plan nennt die ISO 20916 das Clinical Performance Study Protocol (CPSP).
Die ISO 20916:2019 widmet sich in den Anhängen A bis F den besonderen Anforderungen an interventionelle und bestimmte andere risikobehaftete Leistungsstudien gemäß Artikel 58 der IVDR. Ausführliche Vorgaben zu diesen Studien nennt die IVDR im Anhang XIV.
Anhang II: Technische Dokumentation
Der Anhang II beschreibt die Anforderungen an die Technische Dokumentation. In dessen Abschnitt 6 schreibt die IVDR, was die Inhalte der Leistungsbewertung, Verifizierung und Validierung sein sollen.
Anhang VII: Ein Blick hinter die Fassade
Der Anhang VII formuliert die Anforderungen an die Benannten Stellen. Wer versteht, auf was die Benannten Stellen bei den Konformitätsbewertungstätigkeiten achten müssen, kann daraus ableiten, worauf es bei der Leistungsbewertung ankommt und wo die Schnittstellen sind.
Fokus der Prüfungen
Gemäß Anhang VII, Abschnitt 4.5.4 der IVDR soll der Fokus der Prüfung auf den beim Hersteller etablierten Verfahren liegen sowie der Dokumentation zu:
- Planung, Durchführung, Bewertung, Berichterstattung, Aktualisierung der Leistungsbewertung
- Nachbeobachtung der Leistung nach dem Inverkehrbringen (Post-Market Performance Follow-Up, PMPF)
- Schnittstelle zum Risikomanagement
- Beurteilung und Analyse der Daten und ihrer Relevanz in Bezug auf den Nachweis der Konformität mit den grundlegenden Sicherheits- und Leistungsanforderungen gemäß Anhang I der IVDR
- Bericht über die Leistungsbewertung
Der Anhang verpflichtet die Benannten Stellen explizit, Common Specifications, Leitlinien sowie Best-Practice-Guidance-Dokumente zu berücksichtigen.
Literaturrecherchen
Benannte Stellen müssen auch die Ergebnisse der Literaturrecherchen und der durchgeführten Verifizierungen und Validierungen sowie weiterer Tests prüfen. Auch die Verpackung, Stabilitätsstudien und die Ergebnisse der Haltbarkeitsprüfungen müssen sie begutachten.
Die Begutachtung der Leistungsbewertung soll explizit Folgendes umfassen:
- Zweckbestimmung und bestimmungsgemäßer Gebrauch
- Leistungsbewertungsplanung
- Methode und Dokumentation der Literaturrecherche
- Analytische und klinische Leistungsstudien
- PMPF
- Bericht über die Leistungsbewertung
- Etwaige Begründungen, falls Leistungsstudien nicht durchgeführt wurden oder wenn vom vorgegebenen Prozess abgewichen wurde
- Falls zur Leistungsbewertung Daten von als gleichwertig angesehenen Produkten verwendet werden, wird die Validität der angenommenen Gleichwertigkeit und die Eignung der Daten zum Nachweis der Konformität geprüft.
Bemerkenswert ist, dass die Benannte Stelle mit diesen Prüfungen gewährleisten soll, dass
- die „Schlussfolgerungen unter Berücksichtigung des genehmigten Leistungsstudienplans zutreffend sind“,
- die relevanten „Sicherheits- und Leistungsanforderungen gemäß Anhang I angemessen berücksichtigt werden“,
- die Leistungsbewertung „mit den Anforderungen bzgl. des Risikomanagements in Einklang steht“,
- die Leistungsbewertung „gemäß Anhang XIII durchgeführt wird“ und
- sich die Leistungsbewertung „in den zur Verfügung gestellten Produktinformationen auf angemessene Weise widerspiegelt“.
Dies lässt hoffen, dass man zukünftig deutlich mehr Transparenz und Vergleichbarkeit bei Angabe der Leistungsfähigkeit eines IVDs erreicht.
b) MPDG
Das MPDG verweist in Kapitel 4 „Klinische Prüfungen, Leistungsstudien und sonstige klinische Prüfungen“ grundsätzlich auf die Vorgaben der IVDR und ergänzt lediglich besondere nationale Regelungen. Diese betreffen z. B. die Verfahren bei der Ethik-Kommission oder die Beantragung einer Leistungsstudie bei der zuständigen Bundesoberbehörde.
c) MDCG Guidances
Weitere Informationen zur Durchführung und Dokumentation der Leistungsbewertung finden sich in den aktuellen Guidance-Dokumenten der MDCG (Medical Device Coordination Group). Die MDCG-Guidances unterstützen bei der Umsetzung, sind aber nicht rechtlich bindend.
Relevante Dokumente
Hersteller sollten u. a. die folgenden Dokumente beachten:
- MDCG 2022-2 „Guidance on general principles of clinical evidence for In Vitro Diagnostic medical devices (IVDs)“ – Dieser Leitfaden geht auf die grundlegende Vorgehensweise zum klinischen Nachweis basierend auf den drei Elementen der Leistungsbewertung ein: wissenschaftliche Validität, Analyseleistung und klinische Leistung. Das Dokument enthält eine Definition für den „Stand der Technik“, der die essenzielle Basis einer produktspezifischen Leistungsbewertungsstrategie darstellt. Darüber hinaus finden IVD-Hersteller Hinweise zum Umgang mit Leistungsstudien, die sie bereits unter der IVDD durchgeführt haben.
- MDCG 2020-1 „Guidance on clinical evaluation (MDR) / Performance evaluation (IVDR) of medical device software“ – Alle Hersteller von IVD, die sich gemäß MDCG 2019-11 als Medical Device Software (MDSW) qualifizieren, finden in diesem Guidance-Dokument wertvolle Hinweise zur konzeptionellen Umsetzung der Leistungsbewertung für IVD-Software. Dabei sollten sie beachten, dass sich die Definitionen von „wissenschaftlicher Validität“ und „Analyseleistung“ von denen der IVDR unterscheiden, aber für Software viel treffender formuliert sind. Während sich die klinische Bewertung eines Medizinprodukts gemäß MDR sehr von der Leistungsbewertung eines IVDs gemäß IVDR unterscheidet, folgt die klinische Bewertung bzw. Leistungsbewertung von (IVD) MDSW der gleichen strategischen Vorgehensweise. Das ist sehr zu begrüßen, da der klinische Nutzen sowohl einer Medizinprodukte-Software als auch der einer IVD-Software auf der Bereitstellung korrekter, präziser und verlässlicher Informationen liegt. Den Leistungsnachweis daher basierend auf den drei Elementen wissenschaftliche Validität, Analyseleistung und klinische Leistung zu erbringen, ist sinnvoll.
- MDCG 2022-10 „Q&A on the interface between Regulation (EU) 536/2014 on clinical trials for medicinal products for human use (CTR) and Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR)“ – Diese Leitlinie ist insbesondere für IVD-Hersteller und Labore relevant, deren Produkte bzw. Inhouse-IVD während einer klinischen Studie für Arzneimittel Anwendung finden. Sie betrifft aber auch Sponsoren solcher Arzneimittelstudien, die sich die Qualität und Güte der in-vitro-diagnostischen Tests sowie deren Konformität mit den grundlegenden Sicherheits- und Leistungsanforderungen nach Anhang I der IVDR belegen lassen müssen, wenn der Test einen medizinischen Zweck im Zusammenhang mit der Studie erfüllt. Andernfalls gelten die GCP-Anforderungen.
d) FDA
Die FDA hat mehrere Dokumente verfasst, die hier noch ergänzt werden. Sie hat u. a. ein Guidance-Dokument „Biomarker Qualification: Evidentiary Framework“ veröffentlicht.
4. In 8 Schritten zur Leistungsbewertung – wie Sie als Hersteller vorgehen sollten
In diesem Kapitel erfahren Sie, wie Sie Schritt für Schritt vorgehen können, um möglichst zielgerichtet zu gesetzeskonformen Leistungsbewertungen zu gelangen.
1. Schritt: Verfahrensanweisung erstellen und überprüfen
Schreiben Sie zuerst eine Verfahrensanweisung zur Leistungsbewertung von IVD und integrieren Sie diese in Ihr QM-System. Wie Sie zu einer Verfahrensanweisung kommen, erfahren Sie weiter unten.
Achten Sie darauf, dass die Verfahrensanweisung den gesamten Prozess der Leistungsbewertung eines IVDs begleitet. Dieser erstreckt sich über den kompletten Lebenszyklus des Produkts und geht weit über die Entwicklung hinaus.
Hinweis: Zwar fordert die ISO 13485 nicht explizit, einen Prozess zur Leistungsbewertung festzulegen; jedoch fordert sie in Kapitel 4.2.1 e), alle Dokumentationen umzusetzen, die in den regulatorischen Anforderungen genannt werden. In der IVDR findet sich diese Forderung u. a. im Artikel 56 sowie im Anhang XIII. Die IVDR beschreibt die Leistungsbewertung als einen fortlaufenden Prozess. Sie betont die enge Schnittstelle zum Risikomanagement (Anhang VII, Abschnitt 4.5.4 und Anhang XIII, Abschnitt 1.1).
2. Schritt: Produktspezifischen Leistungsbewertungsplan schreiben
Als Nächstes schreiben Sie für jedes IVD einen spezifischen Leistungsbewertungsplan. Beachten Sie dabei, dass Sie alle Inhalte in den Leistungsbewertungsplan aufnehmen, die Anhang XIII, Abschnitt 1.1. der IVDR vorgibt.
Beschreiben Sie im Leistungsbewertungsplan den aktuellen Stand der Technik sowohl in Hinblick auf das medizinisch bzw. diagnostisch relevante Feld als auch in Bezug auf die Technologie.
Recherchieren Sie in Leitlinien, der wissenschaftlichen Fachliteratur sowie in Normen und protokollieren Sie diese Literaturrecherche nachvollziehbar.
Hinweise:
- Aus den Erkenntnissen zum Stand der Technik können Sie als IVD-Hersteller die Produktanforderungen (Entwicklungseingaben gemäß ISO 13485, 7.3.3) und Produktdesignspezifikationen (Entwicklungsergebnisse gemäß ISO 13485, 7.3.4) ableiten.
- Die Recherche nach dem Stand der Technik führt zu Ergebnissen (z. B. über den Nutzen und die Risiken vergleichbarer Produkte), die Sie auch im Risikomanagement benötigen.
- Den Leistungsbewertungsplan erstellen Sie zu Beginn der Entwicklung Ihres IVDs. Es ist üblich, diesen Plan fortlaufend durch neue Erkenntnisse zu ergänzen.
3. Schritt: Wissenschaftliche Validität nachweisen
Nun beginnt die Entwicklung. Weisen Sie möglichst früh im Entwicklungszyklus die wissenschaftliche Validität des bzw. der Analyten nach, die mit dem IVD detektiert bzw. gemessen werden.
Führen Sie hierzu eine weitere systematische Literaturrecherche durch. Dokumentieren Sie auch diese Recherche nachvollziehbar, u. a. die Suchstrategie, die verwendeten Quellen und die Kriterien für die Auswahl der Daten. Auch Stellungnahmen von Fachgesellschaften und Expertengutachten können einbezogen werden.
Falls diese Nachweise nicht ausreichend sind, führen Sie Machbarkeitsstudien durch. Deren Ergebnisse können insbesondere bei neuartigen Analyten zum Nachweis der wissenschaftlichen Validität des Analyten dienen. Ggf. ist zum abschließenden Nachweis der wissenschaftlichen Validität sogar die klinische Leistungsstudie erforderlich.
Fassen Sie alle Ergebnisse in einem Bericht über die wissenschaftliche Validität zusammen. Fügen Sie den Bericht der Leistungsbewertungsakte hinzu, die selbst Teil der Technischen Dokumentation ist.
Hinweis: Nutzen Sie das Protokoll Ihrer Literaturrecherche im fortlaufenden Prozess zur Aktualisierung der Leistungsbewertung sowie während der Nachbeobachtung der Leistung nach dem Inverkehrbringen (PMPF).
4. Schritt: Analytische Leistungsbewertung durchführen
Beginnen Sie nun mit der analytischen Leistungsbewertung als Teil der Verifizierung des IVDs. Beschreiben Sie dazu Ihr geplantes Vorgehen und das konkrete experimentelle Design in einem analytischen Leistungsbewertungsplan (Verifizierungsplan).
Falls Sie ein komplexes IVD-System haben (z. B. bestehend aus IVD-Gerät, IVD-Assay und IVD-Software), planen Sie die Verifizierung
- der einzelnen Subsysteme,
- deren Integration und
- des Gesamtsystems.
Planen Sie nicht nur die Analyse selbst, sondern auch die Probenahme und das Probenhandling genau. Denn nur kontrollierte Bedingungen bei der Entnahme und Vorbereitung der Proben können im Folgenden zu reproduzierbaren und genauen Analyseergebnissen führen.
Führen Sie dann die Bewertung nach Plan durch und fassen Sie die Ergebnisse der analytischen Leistungsstudien abschließend in einem Bericht über die Analyseleistung zusammen. Bewerten Sie in diesem Bericht auch die Ergebnisse.
Hinweise:
- Der Nachweis der Analyseleistung muss immer auf analytischen Leistungsstudien beruhen.
- Die analytische Leistungsbewertung als Teil der Verifizierung dient dem Nachweis der analytischen Leistungsparameter gemäß Anhang I, Abschnitt 9.1 a) der IVDR.
5. Schritt: Stabilität des IVDs nachweisen
Abhängig von der Art Ihres Produkts müssen Sie seine Stabilität nachweisen. Dieser Nachweis ist insbesondere für IVD-Assays bzw. in-vitro-diagnostische Reagenzien von Bedeutung.
Planen Sie zuerst die Stabilitätsstudien. Nutzen Sie dazu die Hilfestellung des Guidance-Dokuments EP25 des Clinical and Laboratory Standards Institute (CLSI). Achten Sie bei der Planung darauf, dass Sie alle Stabilitätsaspekte betrachten, die die IVDR im Anhang II, Abschnitt 6.3 nennt. Dies sind insbesondere:
- Haltbarkeit des Produkts
- Haltbarkeit (z. B. der Reagenzien) nach Anbruch
- Transportstabilität
6. Schritt: Klinische Leistungsbewertung durchführen
Fahren Sie nun mit der Bewertung der klinischen Leistung fort, mit der Sie die diagnostische Genauigkeit Ihres IVDs darlegen.
Hinweise:
- In der Regel benötigen Sie zur Beurteilung der klinischen Leistungsmerkmale klinische Leistungsstudien. In begründeten Ausnahmen können Sie ggf. alleinig auf wissenschaftliche Literatur zurückgreifen (z. B. bei etablierten, standardisierten Tests und gegebenem Nachweis der Gleichwertigkeit der Produkte) oder auf veröffentlichte Erfahrungen, die aus diagnostischen Routinetests gesammelt wurden (s. auch Artikel zu Inhouse-IVD).
- Ihre grundsätzliche Strategie zum Nachweis der klinischen Leistung beschreibt bereits der Leistungsbewertungsplan aus Schritt 2.
Falls Sie Literaturdaten (ergänzend) verwenden, dokumentieren Sie Ihre systematische Literaturrecherche nachvollziehbar.
Schreiben Sie für die klinischen Leistungsstudien erneut einen Plan – das Clinical Performance Study Protocol (CPSP). Achten Sie darauf, dass Sie die Aspekte aufnehmen, die der Anhang XIII, Abschnitt 2 der IVDR sowie die ISO 20916:2019 einfordern. Berücksichtigen Sie zudem, dass der Plan vorsieht, dass alle anwendbaren Parameter erhoben werden, die der Anhang I, Abschnitt 9.1 b) aufführt. Für den Ausschluss von nicht anwendbaren Leistungsmerkmalen müssen Sie eine nachvollziehbare Begründung liefern. Je nach Produktart und Zweckbestimmung können weitere klinische Leistungsmerkmale sinnvoll sein.
Fassen Sie am Schluss die Ergebnisse der klinischen Leistungsbewertung Ihres IVDs in einem Bericht über die klinische Leistung zusammen.
7. Schritt: Bericht zur klinischen Leistungsbewertung erstellen
Fassen Sie abschließend alle Ergebnisse zur wissenschaftlichen Validität, zur analytischen sowie zur klinischen Leistung im Bericht über die Leistungsbewertung zusammen.
Bewerten Sie in dem Bericht den klinischen Nachweis unter Berücksichtigung des aktuellen Stands der Technik und belegen Sie das positive Nutzen-Risiko-Verhältnis Ihres Produkts.
8. Schritt: PMPF-Plan erstellen und umsetzen
Erstellen Sie einen Plan für die Nachbeobachtung der Leistung nach dem Inverkehrbringen, den sogenannten PMPF-Plan. Berücksichtigen Sie dabei die Vorgaben gemäß Anhang XIII, Teil B, Abschnitt 5.2.
Hinweis: PMPF ist das Akronym des englischen Begriffs „Post-Market Performance Follow-up“.
Führen Sie diese Nachbeobachtung für Ihr IVD durch und fassen Sie die Ergebnisse im PMPF-Bericht zusammengefasst. Verwenden Sie die Ergebnisse dieses Berichts, um den Bericht über die Leistungsbewertung bei Bedarf zu aktualisieren.
Hinweis: Bei der Leistungsbewertung handelt es sich um einen fortlaufenden Prozess. Die Aktivitäten zur Leistungsbewertung begleiten den gesamten Lebenszyklus Ihres Produkts. Daher sind Sie als Hersteller verpflichtet, die Leistung Ihres Produkts kontinuierlich zu überwachen und die Leistungsbewertung stets mit verfügbaren neuen Informationen und Daten zu aktualisieren.
5. Was bei der Leistungsbewertung immer wieder schiefläuft
In diesem Kapitel lernen Sie häufige Fehler kennen. Nutzen Sie diese Liste als Checkliste, um unnötigen Ärger und unnötige Kosten zu vermeiden.
a) Unkonkrete Zweckbestimmung
Häufig formulieren Hersteller die Zweckbestimmung umfassend und wenig konkret:
- Fehlende Beschränkung auf Indikation (alle Krebsarten)
- Fehlende Einschränkung der Zielpopulation
Das führt dazu, dass die Leistung ebenfalls ohne Beschränkung z. B. auf Indikationen und Zielpopulationen nachgewiesen werden müssen. Der Umfang der erforderlichen Studien würde dadurch explodieren. Zudem werden Anforderungen, die beispielsweise für Leistungsstudien mit Minderjährigen, Schwangeren oder stillenden Frauen gelten (s. Artikel 61, 62), leicht übersehen.
b) Unsystematisch recherchierter oder dokumentierter Stand der Technik
Wenn Hersteller den Stand der Technik nicht systematisch recherchieren und nicht nachvollziehbar dokumentieren, fehlen
- belastbare Spezifikation von analytischen und klinischen Leistungsdaten,
- die Kenntnis und die Analyse von Wettbewerbsprodukten,
- die Kenntnis der Nutzer, der Nutzungsumgebung und des beabsichtigten klinischen Nutzens. (Was macht der Arzt oder Patient, wenn er die Information des IVDs vorliegen hat? Welche Entscheidung wird basierend auf der Information des IVDs abgeleitet? Welche Handlungsmöglichkeiten und Therapien gibt es?)
Mangelnde Kenntnis des Stands der Technik führt dazu, dass Hersteller Produktanforderungen nicht erheben und beispielsweise keine ausreichenden Kontrollen als Teil des Produkts spezifizieren (z. B. Selbsttests) .
c) Keine dokumentierte systematische Literaturrecherche
Wenn die Literaturrecherche nicht vollständig durchgeführt und nachvollziehbar dokumentiert ist, kann der Hersteller nicht nachweisen, dass er die Leistungsanforderungen erfüllt hat. Das werden Behörden und Benannte Stellen bemerken und die Zulassung verweigern. Darüber hinaus wird die Literaturrecherche während des PMPF- und PMS-Prozesses in der Regel aktualisiert und müsste bei fehlender Systematik und Dokumentation stets neu begonnen werden. Das kostet unnötig Zeit.
d) Unzureichende Pläne für die analytische Leistungsbewertung
Zu den typischen Fehlern zählen fehlende oder unvollständige Pläne für die Leistungsbewertung. Das führt beispielsweise zu analytischen Leistungsstudien ohne ein statistisch valides experimentelles Layout.
Ursache für unzureichende Pläne ist Unkenntnis über den Stand der Technik bei Plänen, der z. B. in den CLSI-Standards beschrieben ist. Auf diese CLSI-Standards verweist übrigens bereits die unter IVDD harmonisierte Norm EN ISO 18113-1:2011 „In vitro diagnostic medical devices – Information supplied by the manufacturer (labelling) – Part 1: Terms, definitions and general requirements„.
e) Ungeeignete Leistungsstudien
Bei den klinischen Leistungsstudien beobachtet man die folgenden Fehler:
- Es gibt keine Herleitung der Fallzahl bzw. der Probenzahl.
- Die Leistungsstudie berücksichtigt nicht ausreichend die Zweckbestimmung. Entsprechend passt das Studiendesign nicht.
- Die klinischen Leistungsparameter leitet der Hersteller nicht aus dem Stand der Technik spezifisch für den versprochenen klinischen Nutzen ab. Damit fehlen korrekte Endpunkte für die Leistungsstudien.
f) Keine Leistungsdaten anhand von Studien
Hersteller hoffen oft, ohne klinische Leistungsdaten anhand von Studien auszukommen. Das ist zwar prinzipiell möglich. Es setzt aber die Gleichwertigkeit zwischen dem zu evaluierendem IVD und dem Vergleichsprodukt voraus. Diese technische und medizinische bzw. diagnostische Gleichwertigkeit muss detailliert analysiert und der Nachweis muss dokumentiert werden. Leider fehlen dazu oft die erforderlichen Informationen.
g) Fehlender Nachweis der wissenschaftlichen Validität
Es genügt nicht, die Analyseleistung nachzuweisen. Vielmehr müssen die Hersteller auch die wissenschaftliche Validität des Analyten darstellen. Das wird vergessen, weil das die IVDD nicht explizit aufgeführt hatte, obwohl es gemäß GHTF/SG5/N7:2012 als Stand der Technik anzusehen wäre.
6. Unterstützung durch das Johner Institut
Die IVD-Experten des Johner Instituts unterstützen IVD-Hersteller bei allen Tätigkeiten und Phasen der Leistungsbewertung:
- Strategieentwicklung
- Ermittlung des Stands der Technik
- Planung und Durchführung der Bewertungen zur
- wissenschaftlichen Validität
- analytischen Leistungsbewertung
- klinischen Leistungsbewertung
- Planung und Durchführung der PMPF-Aktivitäten bzw. -Studien sowie
- Aktualisierung der Leistungsbewertung
Das IVD-Team des Johner Instituts
- hilft beim Erstellen von Verfahrensanweisungen zur Leistungsbewertung.
- führt für Sie Literaturrecherchen durch und unterstützt Sie bei der nachvollziehbaren Dokumentation.
- erstellt für Sie Pläne und Berichte zur Leistungsbewertung.
- unterstützt Sie bei allen Formen der Studien.
- prüft und korrigiert auf Wunsch Ihre Akten vor einer Einreichung.
- führt Seminare, Schulungen und Workshops bei Ihnen vor Ort oder online durch.
Melden Sie sich, wenn Sie rasch zu einer gesetzeskonformen Leistungsbewertung gelangen möchten, die von Ihrer Benannten Stelle problemlos durchgewinkt wird.
7. Fazit & Zusammenfassung
a) Aspekte der Leistungsbewertung
So zentral wie die klinische Bewertung für Medizinproduktehersteller, ist es die Leistungsbewertung für IVD-Hersteller, aber auch für medizinische Labore, die Inhouse-IVD anbieten. Beide setzen auf Daten, die bereits in der Literatur vorhanden sind oder im Rahmen von Studien erhoben werden müssen.
Die Art der Studien und Daten unterscheidet sich deutlich. Bei der Leistungsbewertung von IVD müssen die Hersteller nachweisen:
- Wissenschaftliche Validität
- Analytische Leistungsfähigkeit
- Klinische Leistungsfähigkeit
Für jeden dieser Aspekte bedarf es eigener Pläne, Daten und Bewertungen.
b) Vergleich von IVDD und IVDR
Die Anforderungen an die Leistungsbewertung, die die IVDR und die darin referenzierte ISO 20916:2019 formulieren, sind im Vergleich zur IVDD deutlich umfangreicher geworden. Allerdings galten bereits unter der IVDD die Dokumente der GHTF, der CLSI sowie die EN 13612:2002 als Stand der Technik. Damit gibt es keine nennenswerten Unterschiede.
Das bedeutet, dass IVD-Hersteller, die bereits gemäß IVDD alle relevanten Regularien beachtet hatten, für die IVDR bestens gerüstet sind. Viele IVD werden jedoch mit der IVDR erstmalig einer Konformitätsbewertung durch eine Benannte Stelle unterzogen. Auf die ausführliche Prüfung der Leistungsbewertungsakte sollten sich alle IVD-Hersteller gut vorbereiten.
c) Herausforderung MDR und IVDR?
Herausfordernd ist die Situation für Hersteller, die sowohl die Anforderungen der IVDR an die Leistungsbewertung als auch die Anforderungen der MDR an die klinische Bewertung erfüllen müssen, weil ihre Produkte unter beide Regularien fallen. Dem kann man gegebenenfalls mit einer klugen regulatorischen Strategie begegnen und so die Aufwände verringern.
Haben Sie Fragen zu diesem Artikel? Melden Sie sich einfach über unser Micro-Consulting. Das IVD-Team des Johner Instituts hilft gerne.
Versionshistorie:
- 2022-09-26: Allgemeines Update



Dear Sir/Madam,
I wanted to ask about the calculations of the cut-off value, is it needed for analytical sensitivity and specificity or for diagnostic sensitivity and specificity? One last question please, what does this mean:
This Section shall provide a summary of analytical data with a description of the study design including methods for determining the assay cut-off, such as:
the population(s) studied: demographics, selection, inclusion and exclusion criteria, number of individuals included;
method or mode of characterisation of specimens; and
statistical methods such as Receiver Operator Characteristic (ROC) to generate results and if applicable, define grey-zone/equivocal zone.
how could one formulate this section in the technical documentation?
Thank you again!
Best regards, Lucie
Dear Lucie,
The calculation of a cut-off value may be applied in different contexts of use. For qualitative tests, the cut-off is defined as “the threshold above which the result is reported as positive and below which the result is reported as negative” (source: CLSI Guidance EP12-A2 “User Protocol for Evaluation of Qualitative Test Performance). Accordingly, the cut-off represents the medical decision point. Manufacturers establish the product-specific cut-off value based on the intended purpose of the IVD medical device and the claimed or desired diagnostic sensitivity and diagnostic specificity.
The IVDR requires in Annex II, Section 6.1.2.6. to describe the product-specific approach for assessing the IVD medical device’s cut-off as part of the technical documentation. The detailed information and wording depend on the intended purpose of your IVD medical device and the product-specific performance evaluation strategy. The cut-off is, e.g., dependent on the intended population and may vary with different specimen types. One approach to select the appropriate cut-off is to present diagnostic sensitivity-specificity pairs in a receiver operating characteristic (ROC) plot. Usually, manufacturers estimate the assay cut-off based on a smaller cohort during their analytical performance studies and validate the selected threshold during the clinical performance study based on a statistically validated sample size. The particular requirements for the documentation of the performance evaluation studies are specified in Annex XIII of the IVDR.
Best regards, Catharina
Hallo,
können Sie mir sagen, ob ein Bericht über eine Leistungsstudie auch personenbezogene Daten enthält?
Vielen Dank für eine schnelle Rückmeldung.
Freundliche Grüße,
Gabriele
Liebe Gabriele,
die konkreten Inhalte eines Berichts über die klinische Leistungsstudie sind sehr produkt- und studienspezifisch. Da Hersteller die Studienpopulation gemäß IVDR, Anhang XIII, Abschnitt 2.3.2 m) jedoch im Detail beschreiben müssen, sind in der Regel auch personenbezogene Daten wie Alter und Geschlecht in einem Plan und Bericht über die klinische Leistungsstudie zu finden. Die IVDR fordert „Informationen zu der Population der Leistungsstudie: Spezifizierung der Prüfungsteilnehmer, Auswahlkriterien, Größe der Population der Leistungsstudie, Repräsentativität der Zielpopulation“ im klinischen Leistungsstudienplan zu benennen.
Grundsätzlich müssen Hersteller jede Leistungsstudie im Einklang mit den geltenden Datenschutzvorschriften durchführen (s. IVDR, Artikel 57). Dazu wenden sie u.a. eine Pseudonymisierung der Daten an.
Herzliche Grüße,
Catharina Bertram
Sehr geehrte Frau Dr. Bertram,
Sie erwähnen ein „komplexes IVD-System (z. B. bestehend aus IVD-Gerät, IVD-Assay und IVD-Software).
Wie sieht es aber Ihrer Meinung aus wenn nur ein IVD-Gerät (Analyzer für klinische Chemie) alleine, unabhängig von IVD-assays, als ein Class-A IVD zugelassen wird. Eine wissenschaftliche Validität, analytische- und klinische performance wäre hierbei ja nicht gegeben da die in der IVDR beschriebenen performance characteristics einem Assay zuzuordnen wären.
Könnte man somit den geforderten Nachweis der wissenschaftlichen Validität, analytische- und klinische performance im Zuge der performance evaluation aufgrund Nichtanwendbarkeit „wegargumentieren“ und stattdessen den Analyzer „nur“ einer klassischen Verifizierung&Validierung unterziehen ?
Ich denke da beispielsweise auch an Urinbecher die im Sinne der IVDR ein IVD darstellen. Auch hier würden die Anforderungen an die performance evaluation nicht anwendbar sein.
Mit freundlichen Grüßen,
Anton S.
Sehr geehrter Herr S.,
die Leistungsbewertungsstrategie sollte stets produktspezifisch hergeleitet und begründet werden. Daher lassen sich die Anforderungen an die Leistungsbewertung eines Produkts in der Regel nicht pauschal ausschließen. Dennoch erwähnt die IVDR bereits im Vorwort (65): „Kann mit bestimmten Produkten keine Analyseleistung oder klinische Leistung erzielt werden oder sind bestimmte Leistungsanforderungen nicht anwendbar, sollten im Leistungsbewertungsplan und den dazugehörigen Berichten die mit diesen Anforderungen verbundenen Weglassungen begründet werden.“ Dies greift der Anhang XIII in Abschnitt 1.2.2. und 1.2.3. konkret auf und sagt aus: „Der Hersteller weist die Analyseleistung/klinische Leistung des Produkts anhand aller Parameter gemäß Anhang I Abschnitt 9.1 Buchstabe a/b nach; Parameter, deren Nichtanwendbarkeit begründet werden kann, können jedoch unberücksichtigt bleiben.“ Ein möglicher Ausschluss des Nachweises der wissenschaftlichen Validität wird hingegen nicht erwähnt. Das ist auch logisch, denn wenn die wissenschaftliche Validität eines Produkts nicht gegeben ist, wird es mindestens schwer sein, das nötige positive Nutzen-Risiko-Verhältnis nachzuweisen (s. Anhang I, Abschnitt 1).
Ich stimme Ihnen zu, dass die Definition von wissenschaftlicher Validität, die wir in der IVDR finden, zunächst nicht direkt auf ein Klasse A Gerät anwendbar scheint. Dort heißt es in Artikel 2, Nr. 38: „wissenschaftliche Validität eines Analyten bezeichnet den Zusammenhang eines Analyten mit einem bestimmten klinischen oder physiologischen Zustand“. Der Fokus liegt also auf dem Analyten. Beachten Sie jedoch auch die Anforderungen, um ein Gerät überhaupt als ein Klasse A IVD zu klassifizieren: Gemäß der Klassifizierungsregeln nach Anhang VIII der IVDR, werden entsprechend Regel 5 b) Instrumenten der Klasse A zugeordnet, wenn sie „vom Hersteller speziell für die Verwendung bei In-vitro-Diagnoseverfahren vorgesehen sind“. Artikel 1, Abschnitt 3 a) der IVDR hebt zudem hervor, das Produkte nur unter die IVDR fallen, wenn sie „aufgrund ihrer Merkmale […] speziell für In-vitro-Untersuchungen bestimmt“ sind. Dies wird von der MEDDEV 2.14/1 rev.2 und dem Manual on Borderline and Classification am Beispiel eines Mixers bekräftigt und eine Definition der spezifischen Charakteristika sowie ihres Nachweises im Zusammenhang mit dem/den identifizierten In-vitro-Untersuchungsverfahren gefordert.
Ich empfehle Ihnen daher, die wissenschaftliche Validität für Ihr Produkt in einem Bericht zu belegen. Bei etablierten Produkten lässt sich die wissenschaftliche Validität häufig bereits durch die systematische Literaturrecherche zum Stand der Technik (medizinisch und technologisch) begründen, die ein Teil des Leistungsbewertungsplans ist. Als Hilfestellung empfehle ich Ihnen, die Definition der wissenschaftlichen Validität für Software gemäß Guidance-Dokument MDCG 2020-2 zugrunde zulegen, da sie auf den Output fokussiert und dies auch auf Geräte übertragbar ist.
Zudem sollten Sie in Ihrem Leistungsbewertungsplan gemäß Anhang XIII, Abschnitt 1.1 ihre produktspezifische Leistungsbewertungsstrategie für jeden in Anhang I genannten Leistungsparameter begründen bzw. Akzeptanzkriterien festlegen. Mindestens Parameter wie Reproduzierbarkeit und Wiederholbarkeit sollten für ein Gerät nachgewiesen werden.
Ich hoffe, ich konnte Ihnen mit dieser sehr ausführlichen Antwort weiterhelfen. Sollten Sie weitere Fragen zur Umsetzung Ihrer produktspezifischen Leistungsbewertung haben oder Unterstützung bei den Aktivitäten benötigen, freu ich mich, wenn Sie sich bei mir melden. Wir helfen gern!
Herzliche Grüße,
Catharina Bertram
Dear Sir/Madam,
Is it mandatory to perform the clinical and performance evaluation study in European population by European laboratory for class B and Class C Device according to IVDR? Or the clinical study performed outside Europe in non-European population is also accepted? I would be grateful for your assistance.
Best regards
Milasa
Dear Milasa,
following IVDR, Art. 2, No. 41, clinical performance is defined as: „the ability of a device to yield results that are correlated with a particular clinical condition or a physiological or pathological process or state in accordance with the target population and intended user“. As highlighted, the intended target population shall be considered as specified in the intended purpose of the IVD medical device. As the IVD medical device shall be placed on the EU market under IVDR, the European population would be the focus of the performance studies. If data are obtained from performance studies outside the EU, we recommend critically evaluating and justifying the applicability of such data to demonstrate clinical performance acc. to IVDR. Please note that the clinical performance study plan shall include, among others, „information on the performance study population: specifications of the subjects, selection criteria, size of performance study population, representativity of target population“ (see ISO 20916 and IVDR, Annex XIII, 2.3.2 for details). You may document your product-specific performance evaluation strategy in your performance evaluation plan (see IVDR, Annex XIII, Sec. 1.1). We recommend considering not only the patient characteristics but also the state of the art in medicine, diagnostic practice and technology to substantiate your strategy.
Best regards
Catharina
Sehr geehrte Frau Dr. Bertram,
könnten Sie mir bitte definieren, ab wann ein Test als „etabliert und standardisiert“ zu verstehen ist?
Vielen Dank!
Mit freundlichen Grüßen
Franziska
Liebe Franziska,
die Definition liefert das Guidance Dokument der GHTF „GHTFSG5/N7:2012 – Clinical Evidence for IVD medical devices – Scientific Validity Determination and Performance Evaluation“.
Dort werden „Established and standardized tests“ definiert als:
„These tests have clinical guidelines or consensus for the use of the test, there is more than one commercial test available, and/or international standard or reference materials exist. These tests produce comparable results for the analyte regardless of the method or the manufacturer.“ In dem Guidance Dokument finden Sie auch einige Beispiele genannt.
„Established and non standardized tests“ sind definiert als:
„These tests have clinical guidelines or consensus for the use of the test, and there is more than one commercial test available. While international reference materials may exist, results obtained from different IVD medical devices might not be used interchangeably.“
Es gelten also sehr strikte Anforderungen, die Sie für Ihr zugrundeliegendes IVD z.B. während der Stand-der-Technik-Analyse nachweisen müssten. Darüber hinaus würden Sie detailliert begründen, warum Ihr Produkt in eine der beiden Kategorien fallen würde.