
MDR und IVDR sowie die ISO 13485:2016 formulieren ebenso wie die FDA klare Anforderungen an die Lieferantenbewertung, Lieferantenauswahl und Lieferantenüberwachung.
Dieser Artikel verschafft Ihnen einen Überblick über die regulatorischen Anforderungen an das Lieferantenmanagement. Er gibt zudem Tipps zur praxisnahen Umsetzung und verrät, wann ein Lieferantenaudit notwendig ist.
1. Grundlagen des Lieferantenmanagements
a) Beispiele für Lieferanten und gelieferte Produkte
Sobald Hersteller etwas nicht selbst entwickeln oder produzieren, sondern einkaufen, benötigen sie einen Lieferanten. Zu den Beispielen für extern bereitgestellte Produkte (Waren und Dienstleistungen) zählen:
- Produktentwicklung
Der Hersteller beauftragt, ein komplettes Medizinprodukt zu entwickeln. - Komponentenentwicklung
Der Hersteller beauftragt, einen Teil eines Medizinprodukts zu entwickeln. - Kauf von Komponenten („Katalogware“)
Der Hersteller nutzt ein „fertiges“, d. h. ein bereits bestehendes Produkt innerhalb seines Medizinprodukts. - Kauf oder Miete von Werkzeugen
Der Hersteller kauft oder mietet Produkte als Werkzeuge. Dazu zählt unter anderem „Software as a Service“, z. B. ein Dokumentenmanagementsystem (hier ist zusätzlich die CSV zu beachten). - IT-Services
Der Hersteller nutzt einen IT-Service, z. B. das Hosting eines Servers oder einen Cloud-Service. Hier ist zu unterscheiden, ob dieser Service Teil seiner Produkte wird oder nicht.
b) Lieferantenbewertung, Lieferantenauswahl, Lieferantenüberwachung
Hersteller sollten zuerst Kriterien festlegen, anhand derer sie die potenziellen Lieferanten bewerten. Anschließend führen Sie diese Lieferantenbewertung durch. Auf Basis dieser Lieferantenbewertung wählen sie den oder die geeignetsten Lieferanten aus (Lieferantenauswahl).
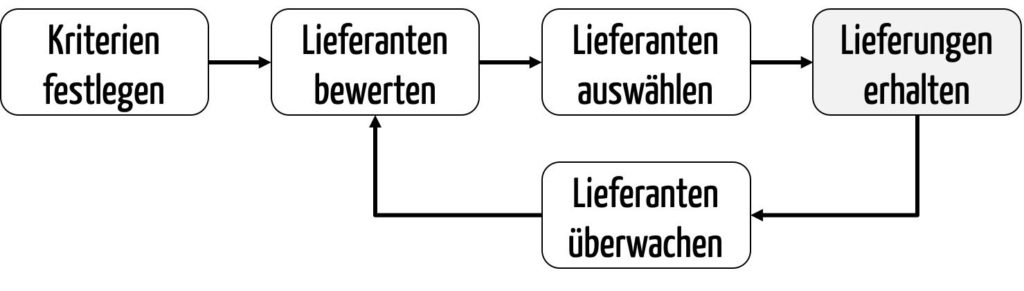
Die Hersteller überwachen die Lieferanten fortlaufend, z. B. im Rahmen von Wareneingangsprüfungen und Lieferantenaudits. Sie bewerten die Lieferanten regelmäßig, etwa anhand der Auditergebnisse und der Qualität der gelieferten Produkte.
Die Kriterien, die man für die initiale Bewertung heranzieht, gleichen meist nicht den Kriterien für die fortlaufende Bewertung im Rahmen der Überwachung. Beispielsweise ist das Kriterium „Liefertreue“ bei der initialen Bewertung nicht messbar, da es noch keine Lieferung gab. Bei der fortlaufenden Bewertung wird die Liefertreue hingegen ein Kriterium sein.
2. Regulatorische Anforderungen an die Steuerung der Lieferanten
a) MDR und IVDR
Anforderungen an das QM-System
MDR und IVDR machen unmissverständlich klar, dass das Qualitätsmanagement „die Auswahl und Kontrolle von Zulieferern und Unterauftragnehmern“ regeln muss (MDR Artikel 10 (9) d) bzw. 10 (8) d) bei der IVDR).
Anforderungen an die Benannte Stelle des Herstellers
Die Benannte Stelle muss entscheiden, ob ein besonderes Audit des Lieferanten oder Unterauftragnehmers notwendig ist (Anhang VII 4.5.2.a, Anhang IX 2.3 und 3.3). Falls dies zutrifft, unterliegen sogar die Lieferanten („Zulieferer“) den unangekündigten Audits – „mindestens einmal alle fünf Jahre“ (Anhang IX 3.4).
Die Benannte Stelle ist verpflichtet, Audits beim Lieferanten durchzuführen, sofern die gelieferten Produkte erheblichen Einfluss auf die Konformität der Medizinprodukte haben und der Hersteller nicht nachweisen kann, dass er die Zulieferer ausreichend kontrolliert (Anhang VII 4.5.2 b)).
Anforderungen an die Technische Dokumentation
Die Hersteller müssen angeben, welche Lieferanten und Unterauftragnehmer bei der Entwicklung und Produktion beteiligt sind (MDR Anhang II, 3. c) bzw. IVDR Anhang II 3.2. b)).
b) ISO 13485 und ISO 9001
Die ISO 9001:2015 und die ISO 13485:2016 stellen konkrete Forderungen an die Auswahl und Bewertung von externen Anbietern von beschafften Produkten. (Die ISO 13485:2016 definiert Produkte als Ergebnisse eines Prozesses und schließt Dienstleistungen in den Produktbegriff mit ein). Nach ISO 13485 müssen Hersteller …
- Kriterien für Lieferanten und die zu beschaffenden Produkte festlegen (Beispiele für Kriterien sind weiter unten genannt),
- Lieferanten gemäß diesen Kriterien beurteilen,
- Lieferanten gemäß diesen Kriterien auswählen,
- Lieferanten gemäß diesen Kriterien überwachen,
- Lieferanten gemäß diesen Kriterien bewerten.
Beachten Sie: Diese Kriterien sind produktspezifisch festzulegen!
Die regulatorischen Anforderungen betreffen neben den Lieferanten auch die Produkte. Hersteller müssen …
- die Auswirkungen des beschafften Produkts auf die Sicherheit und Leistungsfähigkeit des Medizinprodukts analysieren,
- verbundene Risiken adressieren,
- Spezifikationen für die zu beschaffenden Produkte festlegen,
- Anforderungen an Produkte, Qualifikation und Qualitätsmanagement des Lieferanten soweit angemessen benennen,
- festlegen, mit welchen Verfahren, Prozessen und Werkzeugen die gelieferten Produkte zu prüfen sind und
- die Produkte gemäß diesen Vorgaben prüfen.
Die Norm besteht auf schriftlichen Qualitätsvereinbarungen:
Die Lenkungsmaßnahmen müssen in einem angemessenen Verhältnis zu dem damit verbundenen Risiko und der Fähigkeit der externen Parteien […] stehen [und] schriftliche Qualitätsvereinbarungen enthalten.
ISO 13485:2016 Kapitel 4.1.5
c) Vorgaben der ZLG
Weitere Vorgaben zur Lieferantenbewertung finden Sie in den Dokumenten der ZLG, z. B. den Schriftstücken 3.9 B16 und 3.9 B17.
d) Vorgaben der NBOG
Beachten Sie: Die Notified Body Organization Group (NBOG) nennt in ihren NBOG’s Best Practice Guide 2010-1 das alleinige Verlassen auf 9001/13485-Zertifizierungen ausdrücklich als Beispiel mangelnder Kontrolle. Dieses Vorgehen sollte/müsste somit Lieferantenaudits der Benannten Stelle nach sich ziehen.
Der geringe Nutzen von Zertifikaten deckt sich mit den Erfahrungen Benannter Stellen bei diversen Audits: Selbst geschriebene, nicht akkreditierte, unseriöse Zertifikate sind letzten Endes leider nicht mehr als ein Zettel und bieten keine Verlässlichkeit.
Der bessere Ansatz besteht darin, eine gute Qualitätssicherungsvereinbarung (QSV) abzuschließen und ggf. Lieferantenaudits durchzuführen.
e) FDA: 21 CFR part 820
Nahezu identische Anforderungen wie die ISO 13485 nennt die FDA im 21 CFR part 820.50 „Purchasing Controls“. Dies ist z. B. eine Qualitätssicherungsvereinbarung:
Purchasing documents shall include, where possible, an agreement that the suppliers, contractors, and consultants agree to notify the manufacturer of changes in the product or service so that manufacturers may determine whether the changes may affect the quality of a finished device.
FDA 21 CFR part 820.50
Die FDA betont, dass die Daten, die bei der Auswahl, Bewertung und Überwachung der Lieferanten und Produkte entstehen, der Dokumentenlenkung gemäß 21 CFR part 820.40 unterliegen.
3. Lieferantenbewertung praktisch umgesetzt
Wie Sie Ihre Lieferanten auswählen und bewerten, sollten Sie nicht für jeden Einzelfall neu entscheiden, sondern in einer Verfahrensanweisung zur Lieferantenauswahl und Lieferantenbewertung festlegen.

Diese Verfahrensanweisung muss – um die oben genannten Anforderungen zu erfüllen – Kriterien und Methoden für die Lieferantenbewertung und die Lieferantenauswahl bestimmen.
a) Schritt 1: Kriterien festlegen
Wenn Sie die Maßnahmen für die Bewertung und Auswahl Ihrer Lieferanten festlegen, zählen zu den Kriterien, die Sie berücksichtigen können:
- Entwickelt der Lieferant ein Medizinprodukt oder Teile/Komponenten dafür?
- Stellt der Lieferant Dienstleistungen zur Verfügung, die Teil Ihres Produkts werden? Ein Beispiel wäre ein Hosting-Dienstleister, mit dem Sie Ihre Software as a Service anbieten.
- Ist Ihr Lieferant ISO 13485 zertifiziert?
- Wie abhängig sind Sie vom Lieferanten? Gibt es alternative Lieferanten, Produkte oder Verfahren?
- Gibt es bereits Erfahrungen mit dem Lieferanten zur Liefertreue und zur Qualität der gelieferten Produkte?
Eine Google-Suche, die den Lieferanten mit Begriffen wie „Problem“ oder „unzuverlässig“ kombiniert, fördert manchmal neue Erkenntnisse zutage. Auch Produktbewertungen können hilfreich sein. - Ist das Produkt oder die Dienstleistung geschäftskritisch?
Würde eine Nichterfüllung der Anforderungen zu Gesetzesverstößen, zur Verletzung der Datensicherheit, zum Verlust von Firmengeheimnissen, zum Verlust der Reputation oder zu finanziellen Nachteilen führen?
Falls das gelieferte Produkt Software ist oder enthält, sind weitere Kriterien für die Lieferantenbewertung denkbar:
- Welche Sicherheitsklasse hat diese Software?
- Geht es um SOUP bzw. OTS?
- Enthält diese Software selbst SOUP?
- Ist die Software ein Werkzeug oder ein Teil eines Produkts?
- Handelt es sich um Kauf bzw. Miete oder um Entwicklung?
b) Schritt 2: Mögliche Maßnahmen auflisten
Ergreifen Sie abhängig von den Kriterien eine oder mehrere der folgenden Maßnahmen:
- Qualitätssicherungsvereinbarung verhandeln
- Von Ihrem Lieferanten einzuhaltende Normen
- Liste Ihrer Verfahrensanweisungen, die Ihr Lieferant befolgen muss
- Anzahl und Qualifikation des vom Lieferanten bereitzustellenden Personals
- Einverständnis des Lieferanten mit Lieferantenaudits einschließlich Umfang und Frequenz
- Ggf. Einschränkung möglicher Lieferanten auf solche, die ISO-13485-zertifiziert sind
- Wareneingangskontrolle
- Häufigkeit, Stichproben
- Methoden: z. B. Nachtesten, Sichtprüfung
- Art und Umfang der dem Lieferanten bereitgestellten Dokumentation, z. B.
- Produktspezifikationen
- Akzeptanzkriterien
- Projektvorgaben wie Zeit und Budget
- Qualitätssicherungsvereinbarung (s. o.)
- Lieferantenaudit
Lesen Sie hier mehr zum Thema Qualitätssicherungsvereinbarung (QSV), zu den regulatorischen Anforderungen daran und zu den typischen Inhalten einer solchen QSV.
c) Schritt 3: Maßnahmen und Kriterien einander zuordnen
Die eben genannten Methoden und Maßnahmen werden Sie sicher nicht für jeden Lieferanten anwenden. So dürfte ein Audit bei Ihrem Lieferanten für Bürozubehör ein wenig sinnvolles Unterfangen sein. Wenn Ihr Lieferant hingegen die Software für Ihr Medizinprodukt schreibt und nicht selbst ISO-13485-zertifiziert ist, wird ein Lieferantenaudit unverzichtbar sein.
Ein typisches Vorgehen, das zwar nicht gefordert, aber dennoch von den Benannten Stellen gerne gesehen ist (da in NBOG’s Best Practice Guide 2010-1 erwähnt), ist die risikobasierte Einteilung der Lieferanten in kritische Lieferanten und nicht-kritische Lieferanten. Das Dokument der NBOG definiert kritischer Lieferant folgendermaßen:
A critical supplier is a supplier delivering materials, components, or services that may influence the safety and performance of the device.
NBOG BPG 2010-1 2.2
Im letzten Schritt legen Sie daher fest, welche Maßnahmen zur Lieferantenbewertung Sie bei welchen Kriterien anwenden. Da das Regelwerk sehr schnell unübersichtlich werden kann, können Sie die Maßnahmen gruppieren und verschiedene Typen von Lieferanten festlegen.
So könnte es einen Typ „hochkritische Lieferanten“ geben, mit denen Sie eine Qualitätssicherungsvereinbarung unterschreiben, die Audits, eine vollständige Wareneingangsprüfung und Personen einer bestimmten Qualifikationsstufe vorsehen.
Diesen Algorithmus zur Lieferantenbewertung können Sie tabellarisch, textuell oder als Flussdiagramm beschreiben.
4. Lieferantenaudits
Wie oben dargestellt, zählen die Lieferantenaudits zu den Maßnahmen, die Hersteller im Rahmen der initialen Lieferantenbewertung und/oder fortlaufenden Lieferantenüberwachung vornehmen können.
Ob und wann Lieferantenaudits stattfinden müssen, hängt u. a. von der Kritikalität der gelieferten Produkte ab sowie davon, ob die Lieferanten über ein eigenständiges QM-System verfügen.
Mit anderen Worten: Hersteller sollten ihre Lieferanten so gut wie möglich mit Vereinbarungen, Vorgaben und Prüfungen (beim Lieferanten oder Hersteller) lenken. Wenn die Prüfung der Produkte oder Prozesse keine ausreichende Gewissheit verschafft, ist ein Lieferantenaudit angesagt.
a) Lieferantenaudit: Wenn der Lieferant nicht über ein eigenes QM-System verfügt
In diesem Fall erklären die Hersteller ihr eigenes Qualitätsmanagementsystem bzw. die darin formulierten Regeln für ihre Lieferanten als verpflichtend. Sie schreiben dem Lieferanten die Prozesse vor bzw. erarbeiten gemeinsam mit ihm Vorgaben.
Dass sich die Lieferanten an diese Regeln halten, müssen die Hersteller überprüfen; abhängig von der Kritikalität der Produkte zum Beispiel durch Wareneingangskontrollen und ggf. zusätzlichen Lieferantenaudits. Im Rahmen eines solchen Audits kontrollieren die Hersteller etwa, ob der Lieferant die Entwicklung oder Produktion gemäß den Herstellervorgaben dokumentiert.

Die Hersteller selbst werden ebenfalls auditiert. Gemäß ISO 13485 müssen sich diese Audits durch Benannte Stellen auch auf die Lieferanten erstrecken. Es kann also sein, dass der Auditor der Benannten Stelle zum Lieferanten fährt.
Da die Komponentenhersteller und Entwicklungsdienstleister selbst keine Medizinprodukte in den Verkehr bringen, müssten sich diese nicht einem Audit einer Benannten Stelle unterziehen. Sie gestatten das meist nur deshalb, um den Forderungen ihrer Kunden, der Medizinproduktehersteller, gerecht zu werden.
b) Qualitätsmanagementsystem statt Lieferantenaudit
Um dieses „Ausufern“ des eigenen Audits auf die Lieferanten zu vermeiden, bevorzugen viele Hersteller solche Lieferanten, die über ein eigenes QM-System verfügen. In diesem Fall beschränkt sich das Audit durch eine Benannte Stelle normalerweise auf den Hersteller.

Für Hersteller von Medizinprodukten bieten sich bei der Lieferantenauswahl v. a. diejenigen Unternehmen an, die ein Zertifikat nach ISO 13485 und nicht (nur) eines nach ISO 9001 haben.
Die Zertifizierung alleine reicht aber nicht aus: Die Hersteller müssen sicherstellen, dass der „Scope“ des Dienstleister-Zertifikats die Prozesse umfasst, die für den Hersteller relevant sind.
Von einem zusätzlichen Lieferantenaudit soll aber auch bei dieser Variante nicht abgeraten werden. Solche Audits müssten aber als Bestandteil der Verträge zwischen Medizinprodukteherstellern und Lieferanten aufgenommen werden.
c) Welche Firmen man vom Lieferantenaudit ausnehmen kann
Die Konformitätsbewertungsverfahren beziehen sich auf die Entwicklung und Produktion von Medizinprodukten. Immer dann, wenn Hersteller Komponenten für ihre Medizinprodukte entwickeln oder produzieren lassen, können diese Arbeitsschritte Gegenstand eines Lieferantenaudits werden.
Anders sieht es bei Komponenten aus, die nicht speziell für das Medizinprodukt entwickelt oder produziert werden, z. B. Monitore (Bildschirme), Netzteile oder auch „off-the-shelf“ Softwarekomponenten. Hier würden die Hersteller im Rahmen des Risikomanagements sicherstellen, dass diese „Zukaufteile“ („Katalogware“) zu keinen inakzeptablen Risiken führen. Ein Lieferantenaudit würden sie bei Lieferanten solcher Produkte nicht durchführen (dürfen).
Lesen Sie hier mehr zum Thema Audit.
5. Zusammenfassung
a) Lieferantenbewertung und Lieferantenauswahl
- Hersteller müssen Lieferanten vor der Beauftragung bewerten und auswählen. Diese Auswahl muss anhand klarer Kriterien erfolgen.
- Die Steuerung der Lieferanten, d. h. die Lieferantenlenkung – dazu zählt insbesondere auch die Überwachung der Lieferanten – ist ein kontinuierlicher Prozess.
- Die Wahl dieser Kriterien und die Intensität dieser Steuerung müssen risikobasiert erfolgen.
b) Lieferantenaudits
- Lieferantenaudits führt man bei Firmen durch, an die man einen Teil der eigenen Tätigkeiten ausgelagert hat, z. B. einen Teil der Entwicklung. Man spricht hier oft von der „verlängerten Werkbank“. Dann muss das Audit nach den Regeln des QM-Systems des Herstellers (ISO 13485) erfolgen.
- Dieses Audit können sich die Hersteller (Inverkehrbringer) ersparen, wenn der Entwicklungspartner selbst über ein QM-System nach ISO 13485 verfügt und dem Hersteller die entsprechende Dokumentation für das Produkt vorlegt. Das Gleiche gilt für Audits durch die Benannte Stelle.
c) Fazit
Hersteller lagern zunehmend Tätigkeiten wie Entwicklung und Produktion aus, entweder ganz oder in Teilen. Die Regularien machen klar, dass dadurch die Tätigkeiten nicht einem Qualitätsmanagementsystem entzogen werden dürfen. Daher sind die Benannten Stellen verpflichtet, ggf. auch die Lieferanten zu prüfen – u. U. im Rahmen unangekündigter Audits.
Somit sind die Hersteller gut beraten, Lieferanten auszuwählen und zu überwachen, mit denen sie ein durchgängiges Qualitätsmanagement und folglich die Konformität und Sicherheit der Produkte gewährleisten können.
Das Johner Institut unterstützt Hersteller und Lieferanten u. a. bei den folgenden Tätigkeiten:
- Verfahrensanweisungen für die Bewertung, Auswahl und Überwachung von Lieferanten erstellen, die konform sind mit MDR, IVDR, ISO13485 und FDA
- Qualitätssicherungsvereinbarungen formulieren
- Audits durch Hersteller und Benannte Stellen vorbereiten
- Lieferantenaudits im Namen des Herstellers durchführen
- Korrektes Zusammenspiel der Tätigkeiten (z. B. Lieferantenüberwachung, Risikomanagement, Trendanalyse etc.) im Rahmen der Post-Market-Surveillance (zentrale Anforderung von MDR und IVDR) gewährleisten
- Übernahme der Rolle des externen QM-Beauftragten
Nehmen Sie gleich Kontakt mit uns auf!
Versionshistorie
- 2025-01-08:
- Redaktionelle Änderungen
- Kapitel 3. c) erweitert um einen Absatz zu „kritische Lieferanten“
- Kapitel 4 präzisiert zur Durchführung von Lieferantenaudits
- 2023-05-31: Redaktionelle Änderungen



Eine Anmerkung zu Zertifizierungen: interessanterweise nennt „NBOG’s Best Practice Guide 2010-1“ das alleinige Verlassen auf 9001/13485-Zertifizierungen ausdrücklich als Beispiel *mangelnder* Kontrolle. Dieses Vorgehen sollte/müsste somit Lieferantenaudits der Benannten Stelle nach sich ziehen.
Der geringe Nutzen von Zertifikaten deckt sich mit diversen Auditerfahrungen zu selbst geschriebenen, nicht akkreditierten, unseriösen Zertifikaten, die letzten Endes leider nicht mehr als ein Zettel sind und keine Verlässlichkeit bieten.
Dann doch lieber eine gute QSV und Lieferantenaudits!
Super Hinweis! Danke, lieber Jan! Das ergänze ich sofort! Liebe Grüße, Christian
Sehr geehrter Herr Johner
Sie schreiben in diesem Artikel:
„Da die Komponentenhersteller und Entwicklungsdienstleister selbst keine Medizinprodukte in den Verkehr bringen, müssten sich diese überhaupt keinem Audit einer benannten Stelle unterziehen.“
Gilt dies auch nach der MDR?
Mit freundlichen Grüssen
A. Mahlau
Sehr geehrter Herr Mahlau,
so ist es! Daran ändert die MDR nichts.
Allerdings zwischen viele Medizinproduktehersteller ihre Dienstleister und Komponentenhersteller dazu, sich zertifizieren zu lassen. So etwas ist seid ISO 13485:2016 auch möglich.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
Vielen Dank für Ihren Überblick. Unter Abschnitt 2.a) verweisen Sie auf Anhang VII 5.5.2, welcher nicht existiert. Handelt es sich hierbei um einen Tippfehler?
Mit Freundlichen Grüßen
Carolin Isenberg
Sehr geehrte Frau Isenberg,
danke für Ihre scharfen Augen! Sie haben Recht! Es müsste nicht „5.5.2“ lauten, sondern „4.5.2“. Dort, kurz oberhalb von Abschnitt 4.5.2 findet sich die Forderung.
Danke, dass Sie diesen Fehler entdeckt und mir damit die Möglichkeit gegeben haben, diesen zu beheben!
Beste Grüße, Christian Johner
Sehr geehrter Prof. Johner,
hat die benannte Stelle auch bei ISO 13485 zertifizierten Klasse I-Herstellern das Recht die Lieferanten zu auditieren? Annex VII und IX kommen im Rahmen von Konformitätsbewertungstätigkeiten zur Anwendung. Diese finden jedoch erst ab Klasse I* in Zusammenarbeit mit benannten Stellen statt. Oder gibt es noch eine andere Rechtsgrundlage bspw. aufgrund der ISO 13485, weshalb Lieferanten von Klasse I-Herstellern von Audits der benannten Stelle betroffen sein könnten?
Mit freundlichen Grüßen
M. Tschirner
Sehr geehrte Frau Tschirner,
eine Benannte Stelle hat zuerst einmal überhaupt kein Recht, einen Lieferanten zu auditieren. Schließlich hat sie mit diesem Lieferanten keinen Vertrag. Eine Benannte Stelle ist auch keine Polizeibehörde mit Durchsuchungsgewalt.
Es ist eher die Aufgabe des Inverkehrbringers sicherzustellen, dass die Benannte Stelle die Konformität des Herstellers (z.B. QM-System) und seiner Produkte beurteilen kann. Wenn der Hersteller das nicht gewährleistet, muss die Benannte Stelle ggf. die Zertifizierung ablehnen.
D.h. es könnte für den Hersteller wichtig sein, dass die Benannte Stelle die Möglichkeit hat, den Hersteller zu auditieren.
Wenn es allerdings soweit kommt, ist möglicherweise etwas anderes schiefgelaufen. z.B. hat der Hersteller es versäumt, Audits bei seinem Lieferanten zu machen, oder er hat Dinge ausgelagert, die er besser nicht ausgelagert hätte, oder er hat keine Möglichkeit geschaffen, um die gelieferten Gegenstände ausreichend einer „Wareneingangskontrolle“ zu unterziehen, oder er hat keinen Lieferanten ausgewählt, der bereits zertifiziert ist.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
Im Zusammenhang von ISO13485 und IVDR (3.2b => Information zur Herstellung: Angabe aller Stellen, einschließlich Lieferanten und Unterauftragnehmer, bei denen Herstellungstätigkeiten durchgeführt werden.) ist mir folgender Sachverhalt unklar:
Muss man als Auftragsfertiger dem Hersteller eine Liste der Zulieferer ausfertigen und wenn ja, gilt das für alle Zulieferer oder ist dies auf Zulieferer für „kritische Komponenten“ begrenzt? Oder muss der Hersteller nur den Namen des Auftragsfertiger (in diesem Fall dann Lieferant gegenüber Hersteller) bei der benannten Stelle benennen?
Mit freundlichen Grüßen
S. Plasonig
Sehr geehrter Herr Plasonig,
als Auftragsfertiger, d.h. als „Nicht-Inverkehrbringen“, müssen Sie aus Sicht des Medizinprodukterechts erst einmal gar nichts machen. Es ist der Inverkehrbringer sprich der Hersteller, der Ihnen gewisse Pflichten auferlegen sollte, um seine eigenen Pflichten zu erfüllen.
Aus Herstellersicht macht es Sinn, darauf zu bestehen, dass der Auftragsfertiger bei allen Änderungen informiert, die sich auf die Konformität, Sicherheit und Leistungsfähigkeit des Produkts auswirken können. Das bezieht sich au die Lieferanten des Auftragsfertigers.
Da viele Auftragsfertiger diese Unterscheidung nicht einfach machen können, verlangen die Hersteller die Liste aller Zulieferer.
Mir ist nicht ganz klar, auf welchen „3.2b“ sie sich beziehen.
Generell sollten Überprüfungen immer risikobasiert erfolgen, was voraussetzt, dass man die Risiken beurteilen kann.
Viele Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
die ISO13485 schreibt eine Lieferantenbewertung vor. Betrifft diese aber nicht eigentlich nur diese Lieferanten, die Produkte liefern, die Teil des Medizinprodukts bzw. zu dessen Herstellung (Werkzeuge) oder Qualitätskontrolle notwendig sind?
Warum muss ich die Lieferanten eines PC-Bildschirms, eines Tisches oder weiterem Bürobedarf überhaupt bewerten? Diese Dinge haben doch keinerlei Einfluss auf meine Medizinprodukte.
Wir haben hierzu pauschal in unserer Verfahrensanweisung abgelehnt einen Lieferantenbewertung für Büromaterial und Inventar durchzuführen. Da von diesen Dingen kein Risiko ausgeht, dürfte dies doch in dieser Form möglich sein?
Lieber Herr Handt,
vielen Dank für Ihren Kommentar.
Die ISO 13485 gibt in Kapitel 7.4.1 vor, dass die von Ihnen festgelegten Kriterien für die Auswahl Ihrer Lieferanten auf den Einfluss der Produkte des Lieferanten auf Ihr Medizinprodukt und dem mit dem Medizinprodukt verbundenen Risiko beruhen müssen.
Dementsprechend sollten Sie vor einer Beschaffung, den Einfluss des zu beschaffenden Produktes auf Ihr Medizinprodukt ermitteln. Hat nun das zu beschaffende Produkt keinen Einfluss auf Ihr Medizinprodukt, was wahrscheinlich in den allermeisten Fällen auf z. B. Bürostühle zutrifft, kann begründet werden, dass keine Kriterien für die Auswahl von Lieferanten von Bürostühlen festgelegt werden müssen. Somit gibt es auch keine Anforderungen, die überwacht werden müssen und demnach müssen Lieferanten von Bürostühlen auch nicht wiederbewertet werden.
Ihr vorgehen, bestimmte Produkte (Bürobedarf) und damit deren Lieferanten von der Überwachung pauschal auszunehmen kann demnach durchaus vertretbar sein, wenn es entsprechend begründet werden kann. Dennoch sollte man bei der Beschaffung eines neuen Produktes, auch wenn man einen etablierten Lieferanten beauftragt, prüfen ob dieses einen Einfluss auf das Mediziniprodukt hat und entsprechend des Ergebnisses handeln.