
Mit dem 21 CFR Part 11 formuliert die FDA ihre Anforderungen an die elektronischen Aufzeichnungen und Unterschriften. Diese Anforderungen richten sich auch an Medizinproduktehersteller.
Viele Firmen drucken Dokumente auf Papier aus und unterschreiben diese dann mit Hand, um die Anforderungen des „Part 11“ zu umgehen. Andere nutzen Signatur-Dienstleister. Doch ist das alles notwendig?
1. 21 CFR Part 11: Ein Angstgespenst!?
Die FDA hat mit dem im Jahr 1993 erschienen Part 11 zu den Electronic Records; Electronic Signatures“ v. a. vielen Firmen im Pharmaumfeld schlaflose Nächte bereitet – und den Beratern gute Geschäfte.
Manchmal wurden die Anforderungen so übertrieben interpretiert, dass sich die FDA im Jahr 2003 zu Klarstellungen bemüßigt fühlte. Veröffentlicht hat sie diese z. B. im Guidance-Dokument „Part 11, Electronic Records; Electronic Signatures — Scope and Application“. Schließlich sah sie die eigene Zielstellung konterkariert, nämlich eine Grundlage für den Ersatz von Papierdokumenten durch elektronische Informationen zu ermöglichen.
Doch was fordert der 21 CFR Part 11 wirklich? Und welche Dokumente sind betroffen?
2. Betroffene Dokumente und Systeme
a) Betroffene Dokumente / Aufzeichnungen
Der 21 CFR Part 11 findet immer dann Anwendung, wenn Informationen elektronisch erzeugt, verändert, gespeichert, übertragen oder auf diese zugegriffen werden sollen. Dabei kann es sich um verschiedene Typen an Informationen handeln, beispielsweise
- Texte,
- Bilder, Videos oder
- Audiodateien.
Die Anforderungen aus 21 CRF Part 11 (an die IT-Systeme) sind zu erfüllen, wenn die damit erzeugten, gespeicherten, übertragenen oder veränderten Dokumente dazu dienen, die Einhaltung regulatorischer Vorschriften nachzuweisen. Beispiele dafür sind:
- Freigabe- und Testprotokolle
- Verfahrens- und Arbeitsanweisungen
- Konstruktionszeichnungen, Dokumentation der Software-Architektur
- Spezifikationen, Anforderungsdokumente
- Aufzeichnungen, z. B. aus der Produktion
- Review-Protokolle
- Ausgefüllte Checklisten und Formulare
Als Faustformel kann man sagen, dass Systeme dann dem 21 CFR Part 11 unterliegen, wenn die damit „verwalteten“ Dokumente
- bei der FDA eingereicht werden (z. B. bei einer 510(k)-Submission) oder
- bei einer FDA-Inspektion relevant sind, d. h. die Prüfung des QM-Systems konform 21 CFR Part 820 zum Ziel hat.
Die FDA verzichtet bei folgenden Systemen darauf, dass sie „part 11 compliant“ erstellt werden:
- Alt-Systeme, die vor dem 20. August 1997 in Betrieb waren
- Systeme, die Papierausdrucke erzeugen
Der 21 CFR Part 11 ist also nur dann anzuwenden, wenn elektronische Aufzeichnungen das Papier ersetzen.
Es liegt ein Graubereich vor, wenn ein System zwar einen Papierausdruck erzeugt, man sich bei dessen Erstellung aber auf die elektronische Aufzeichnung verlässt. Beispielsweise erzeugen Hersteller oft automatisiert Tausende Seiten an Testberichten, drucken sie aus und unterschreiben. Hier müsste man die Entscheidung für die Nichtanwendung des 21 CFR Part 11 begründen.
b) Betroffene Systeme
Computerisierte Systeme, welche diese Dokumente erzeugen, müssen den gesetzlichen Anforderungen genügen, beispielsweise dokumentiert und validiert sein. Beispiele für solche Systeme können sein:
- Test- und Prüfstände
- Software-Tools zur Bestimmung von Code-Metriken und zur Ausführung von automatisierten Tests
- Tools wie Polarion, Jira und Confluence, mit denen Aufzeichnungen erstellt und z. B. Checklisten ausgefüllt werden
- Webbasierte Software, mit der Dokumente (z. B. Spezifikationen) erstellt werden
- Produktions- und Prozessanlagen, welche automatisiert protokollieren
3. Die Anforderungen des 21 CFR Part 11
Der 21 CFR Part 11 umfasst drei „Subpart“ (s. Abb. 1):
- Subpart A: Allgemeine Festlegungen, insbesondere Definitionen
- Subpart B: Anforderungen an elektronische Aufzeichnungen
- Subpart C: Anforderungen an elektronische Unterschriften
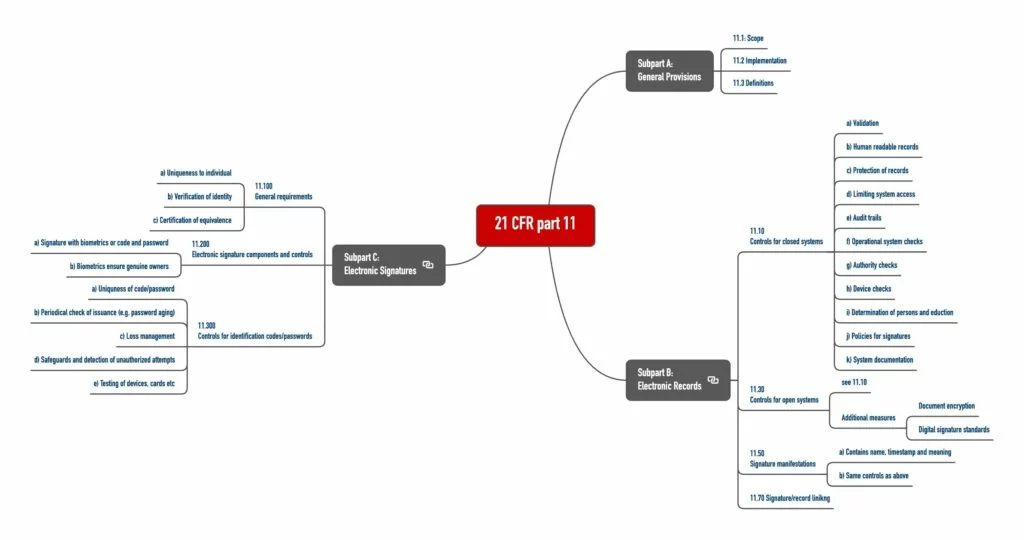
Die Anforderungen der FDA unterscheiden offene und geschlossene Systeme. Ein System ist dann geschlossen, wenn das System unter der Kontrolle von Personen ist, die für die elektronischen Aufzeichnungen verantwortlich sind, die durch dieses System verwaltet werden. Andernfalls ist es ein offenes System.
- Ein Beispiel für ein geschlossenes System ist ein Build- und Test-System im Intranet, auf das nur die verantwortlichen Tester oder Entwickler zugreifen können.
- Ein System, das Daten über das Internet überträgt, zählt zu den offenen Systemen.
a) Anforderungen an geschlossene Systeme
Der 21 CFR Part 11.10 legt die Anforderungen an geschlossene Systeme fest. Personen, die mit diesen Systemen arbeiten, müssen die Authentizität, Integrität und ggf. die Vertraulichkeit der Daten sicherstellen. Dafür sind sie zu Folgendem verpflichtet:
- System validieren (Performanz, Fähigkeit, Veränderungen an Aufzeichnungen oder ungültige Aufzeichnungen erkennen)
- (Auch) Menschenlesbare Aufzeichnungen erzeugen
- Schutz der Aufzeichnungen sicherstellen (Aufzeichnungen müssen verfügbar sein.)
- Begrenzung des Zugriffs auf autorisierte Personen
- Audit-Trails, die computergeneriert sind, müssen Zeitstempel enthalten und zeigen, wer wann was geändert hat. Hier rudert die FDA aber zurück, wie Sie im o. g. Guidance-Dokument nachlesen können.
- Verfahrensprüfungen, um sicherzustellen, dass (nur) die zugelassene Reihenfolge von Arbeitsschritten erzwungen wird – soweit notwendig
- Zugangsprüfungen, um sicherzustellen, dass nur berechtigte Nutzer das System nutzen (z. B. Dokumente erzeugen und unterschreiben) sowie auf das Betriebssystem, den Computer oder die Peripherie zugreifen können
- Prüfung der Peripherie, um sicherzustellen, dass die Ein- und Ausgaben korrekt sind
- Ausbildung der Personen, die mit dem System arbeiten oder es entwickeln
- Verhindern von Fälschungen dadurch, dass man Personen schriftlich haftbar für das macht, was sie unterschreiben.
- Systemdokumentation, z. B. darüber, wer Zugang zum System hat, wie dieser Zugang gewährt wird (sei es für die Anwendung oder Pflege des Systems) sowie wer wann was am System geändert hat
Die FDA fordert, dass die oben diskutierten IT-Systeme validiert werden müssen, und verweist in diesem Zusammenhang auf das Guidance-Dokument „General Principals of Software Validation“. Das wirft die Frage auf, ob es hier nur um die Validierung geht oder um den kompletten Software-Lebenszyklus. Es ist letztes gemeint.
Lesen Sie hier mehr zu diesem Thema:
- Was versteht man unter Computerized System Validation?
- Was ist eigentlich Software Validation?
b) Anforderungen an offene Systeme
An offene Systeme stellt der 21 CFR Part 11.30 zusätzliche Anforderungen. Dazu zählen Maßnahmen wie die Verschlüsselung von Dokumenten und digitale Unterschriftenstandards, um die Echtheit, Integrität und Vertraulichkeit von Aufzeichnungen sicherzustellen.
c) Anforderungen an digitale Unterschriften
Die Anforderungen des 21 CFR Part 11 an digitale Unterschriften werden jedem all jenen vorkommen, die sich mit diesem Thema und z. B. dem Signaturgesetz auseinandergesetzt haben:
- Inhalte: Die digitale Unterschrift muss enthalten
- den Namen des Unterzeichnenden,
- das Datum und die Zeit der Unterzeichnung und
- die Bedeutung der Unterzeichnung (z. B. Review, Genehmigung, Autor).
- Verfälschungssicherheit: Die digitale Unterschrift darf nicht verfälscht werden können. (De facto stellt der 21 CFR die gleichen Anforderungen wie an die Dokumente.)
- Verbindung mit Dokument: Die Unterschrift muss mit dem Dokument so verbunden sein, dass sie nicht auf andere Dokumente angewendet werden kann.
- Einzigartigkeit: Natürlich muss die Unterschrift einem einzelnen Individuum zugeordnet werden können.
- Biometrische und nicht biometrische Verfahren: Die Identifizierung muss auf biometrischen Verfahren beruhen oder aus zwei Komponenten wie Identifizierungscode und Passwort bestehen.
Auch an die Verwendung von Identifizierungscode (z. B. Benutzername, Kürzel oder Nummer) und Passwörter stellt der 21 CFR Part 11 Anforderungen, konkret in 11.200 (a) und 11.300:
- Vier-Augen-Prinzip: Die Vergabe muss so geregelt sein, dass der missbräuchliche Versuch, die Unterschrift eines anderen zu nutzen, zumindest zweier Personen bedürfte.
- Eindeutige Kombinationen: Eine doppelte Vergabe von Codes und Passwörtern muss ausgeschlossen sein.
- Aktualisierung: Beide müssen regelmäßig überprüft werden, ob sie noch ausreichend sicher sind.
- Verlust-Management: Für den Verlust von Codes, Passwörtern, Karten u. ä. muss es ein Verfahren geben, das z. B. die „De-Autorisierung“ gestattet.
- Schutzmaßnahmen: Es müssen geeignete Maßnahmen zum Schutz und zur Detektion von unberechtigten Zugriffsversuchen getroffen sein.
- Überprüfung: Regelmäßig sind die Ein-/Ausgabegeräte einschließlich Karten, die Autorisierungsinformationen tragen oder lesen, auf korrekte Funktionalität zu prüfen.
4. Häufige Fragen im Kontext des 21 CFR Part 11
a) Gibt es Lösungen, um 21 CFR Part 11-Compliance zu gewährleisten?
Die klare Antwort heißt „nein“. Denn der 21 CFR Part 11 verlangt nicht nur technische, sondern auch organisatorische Maßnahmen. Die können Sie nicht kaufen.
Hersteller wie unsere Schwesterfirma Medsoto haben allerdings die Produkte so vorbereitet, dass die technischen Voraussetzungen gegeben sind, um eine (technische) Dokumentation zu erstellen.
b) Muss man den 21 CFR Part 11 einhalten, wenn man alles ausdruckt und dann unterschreibt?
Die Antwort, die in den meisten Fällen zutrifft, lautet „nein“. Es gibt allerdings Ausnahmen, die wir weiter oben bereits am Beispiel einer Testdokumentation angesprochen haben.
c) Muss man überhaupt auf Papier verzichten?
Die FDA verlangt (zunehmend), dass Sie Ihre Unterlagen elektronisch einreichen. Ein Beispiel ist das eStar-Format. Allerdings könnten Sie auch Ausdrucke einscannen und einreichen. Damit könnten Sie von der o.g. Ausnahme abgesehen den Part 11 ignorieren.
d) Was mache ich, wenn ich keine Dokumente, sondern strukturierte Daten habe?
Immer mehr Hersteller, Behörden und Benannte Stellen verstehen die Vorteile, die strukturierte Daten gegenüber Dokumenten haben. Dazu zählen:
- Redundanzfreiheit und damit keine Inkonsistenzen
- Automatische Prüfbarkeit durch Algorithmen
- Flexibilität, die Daten in verschiedene Zielformate für internationale Behörden und Benannte Stellen zu exportieren
Für diese strukturierten Daten gelten die gleichen Anforderungen, beispielsweise:
- Validierung der Systeme
- Lesbarkeit der Daten
- Berechtigungskonzepte und Vorgaben für die Freigaben
- Schutz der Daten
- Unterschriften
Diese Anforderungen können erfüllt werden, ohne dass klassische Dokumente wie Word-Dateien oder PDFs erstellt werden. Der 21 CFR Part 11 verhindert also nicht das Arbeiten mit strukturierten Daten.
(Strukturierte) Daten, die Hersteller, Behörden und Benannte Stellen austauschen, sind serialisiert, beispielsweise im JSON- oder XML-Format. Diese Datenströme entsprechen wiederum Dokumenten, welche den Anforderungen des 21 CFR Part 11 genügen müssen, also lesbar sind oder Änderungen erkennen lassen.
e) Wie kann man die Anforderungen an elektronische Unterschriften am besten erfüllen?
Eingescannte Unterschrift: Das genügt nicht
Ein erster Gedanke könnte sein, eine Unterschrift einzuscannen, in das Dokument einzufügen und jenes als PDF zu drucken. Doch das würde der Forderung der Part 11.70 nicht genügen. Schließlich könnte man diese Grafik als Screenshot exportieren und in ein anderes Dokument einfügen.
Verwendung geeigneter Systeme: Meist der Königsweg
Vielmehr wird üblicherweise eine Prüfziffer des Dokuments (Hashcode) mit dem privaten Schlüssel des Unterzeichnenden verschlüsselt. Dieser verschlüsselte Hashwert ist die digitale Signatur/Unterschrift.
Den Hashcode müssen die computerisierten Systeme (beispielsweise Dokumentenmanagementsysteme) erzeugen können. (Das geschieht für die Anwender nicht wahrnehmbar.)
Die FDA stellt im 21 CFR Part 11 keine Forderungen nach einer qualifizierten Signatur. Das bedeutet, dass eine fortgeschrittene Signatur in den meisten Fällen ausreichen wird.
Der Artikel zu den elektronischen Unterschriften stellt die verschiedenen Signatur-Levels vor.
Signatur-Dienstleister: Konform, aber mit Nachteilen behaftet
Andere Hersteller nutzen Dienstleister, um Dokumente zu signieren. Das hat allerdings auch Nachteile:
- Diese Dienste kosten (bei jeder Unterschrift) Geld.
- Der Workflow wird dadurch regelmäßig gebremst.
- Die Dienstleister setzen auf Dokumente. Deren Konzept harmoniert nicht gut mit dem der strukturierten Daten.
Der Artikel zu den elektronischen Unterschriften und digitalen Signaturen gibt weiterführende Tipps und erläutert, welche Signatur-Stufe üblicherweise benötigt wird.
5. Fazit
Die Anforderungen des 21 CFR Part 11 sind vergleichbar mit den Anforderungen, die Medizinproduktehersteller in Europa erfüllen müssen. Einige zusätzliche Anforderungen wie jene an die Audit-Trails müssen jedoch beachtet werden. Allerdings ist die FDA mit ihrem Guidance-Dokument genau in diesen Bereichen wieder etwas zurückgerudert.
Eine fortgeschrittene elektronische Signatur erfüllt die Anforderungen der FDA an elektronische Unterschriften. Dieses Signatur-Niveau können die Hersteller von Medizinprodukten und Software auch ohne externe „Vertrauensdiensteanbieter“ erreichen.
Auch bei elektronischen Aufzeichnungen und Unterschriften sollte man den gesunden Menschenverstand walten lassen: Daten dürfen nicht unbemerkt verändert werden, insbesondere wenn sie bereits freigegeben wurden. Und man will wissen, wer diese Daten wann freigegeben hat, um die Konformität von Prozessen bewerten zu können.
Um mehr geht es der FDA im 21 CFR Part 11 gar nicht.
Änderungshistorie
- 2024-08-06: Artikel weitgehend überarbeitet und strukturiert, Grafik ausgetauscht
- 2015-11-13: Erste Version des Artikels



Hallo,
die Frage und Antwort habe ich nicht ganz verstanden, Zitat:
„Muss man den 21 CFR part 11 einhalten, wenn man alles ausdruckt und dann unterschreibt?Die Antwort, die in den meisten Fällen zutrifft, lautet „ja“. Es gibt allerdings Ausnahmen, die wir weiter oben bereits am Beispiel einer Testdokumentation angesprochen haben.“ Ich verstehe es so, dass man zwar immer noch eine (Software-)Validierung zeigen muss, man mit einem „kompletten“ Ausdruck aber den Part 11 nicht anwenden müsste, weil ja alle Electronic Records als Hardcopy vorliegen. Wobei der Begriff „komplett ausgedruckt“ häufig ähnlich naiv ist wie die Feststellung „unsere Software ist fehlerfrei“. Also Vorsicht.
Aktuell werden aber andere Fragestellungen diskutiert, nämlich wie man seine Datenintegrität (elektronisch oder Papier, siehe dazu auch das MHRA-Papier vom März 2015) gewährleistet und einem Inspektor demonstriert. „Datenintegrität“ war das Thema für uns und den Inspektionen der FDA und der deutschen (Pharma-)Behördenaufsichten.
Was ich versuchte zu schreiben war:
Wenn Sie etwas ausdrucken und unterschreiben, das man überblicken kann, dann handelt es sich nicht um ein Dokument, das unter den part 11 fällt.
Wenn Sie aber automatisiert eine mehrere 1000 Seiten umfassende und automatische generierte Dokumentation erzeugen, ausdrucken und unterschreiben, dann wird man in Abrede stellen, dass Sie das Dokument wirklich geprüft haben. Das sieht die FDA weiter als ein elektronisches Dokument.
Man hat in diesem zweiten Falls die IT nicht wie einen Word-Processor genutzt, um etwas zu schreiben (sozusagen als Schreibmaschine), sondern als automatisierten Dokumenten-Generator. Das unterscheidet die FDA.
Hallo,
wir sind gerade am diskutieren, wie Compiler oder Testautomisierungswerkzeuge unter Part 11 zu verstehen sind.
Fallen die Werkzeuge, weil sie z.B. Softwarebuilds, Buildreports/protokolle, Testergebnisse (Unit Test Reports oder von automatisierten Tests) erzeugen unter „Part 11“? Bzw. wie fallen sie darunter.
Die Testergebnisse der Unit Tests und automatisierten Tests werden bei uns bereits im Buildsystem geprüft, wir akzeptieren nur Builds, wo alle diese Tests ohne Fehler durchlaufen. Die Ergebnisse aus diesen Tests werden relativ detailliert vom Buildsystem in „druckbare“ Testprotokolle überführt (die entsprechenden Testfälle sind bei uns Teil der Traceability). Die Testprotokolle sind gelenkte Dokumente, die zwar nicht 1000 Seiten haben, aber da diese automatisiert erzeugt wurde, dies die FDA aus unserer Sicht als elektronisches Dokument ansehen würde. Verwaltet werden die gelenkten Dokumente bei uns mit einem Versionskontrollsystem.
Alle einzelnen Werkzeuge haben wir validiert, nur mit den oben genannten Punkten tun wir uns schwer, die auf die einzelnen Werkzeuge anzuwenden. Für z.B. die automatisierten Tests können wir das „System validieren“ und auch dass es „menschenlesbare Aufzeichnungen“ erzeugt. Die meisten restlichen Punkte „Schutz der Aufzeichnungen“, „Zugangsprüfungen“ werden durch die gelenkten Dokumente im Versionskontrollsystem sichergestellt.
Auf Ihrer Seite zum Thema „Unit Testing und IEC 62304“ (https://www.johner-institut.de/blog/iec-62304-medizinische-software/unit-testing-iec-62304/) schreiben Sie, dass Sie die Ergebnisse der Unit Tests unter Versionskontrolle stellen und die Bewertung und Freigabe im Rahmen des Software-Releases formell machen. Wie haben Sie diesen Prozess im Sinne von „Part 11“ dokumentiert?
Sehr geehrter Herr Hermannsen,
danke für die spannenden Fragen. Hier einige Gedanken dazu:
Der Part 11 bezieht sich auf „electronic records und signatures“. Ein Compiler sollte daher nicht in den Scope der part 11 fallen. Bei einem Testautomatisierungstool kann ich das nicht sagen, weil ich nicht weiß, ob die Spezifikationen bzw. Ergebnisse ausschließlich elektronisch vorliegen.
Unabhängig davon sind Werkzeuge aber ggf. zu validieren, was Sie ja tun.
Wir checken einige Build-Artefakte automatisch ins Versionsverwaltungssystem ein. Dazu zählen auch die Testergebnisse. Die eigentliche Freigabe kann aber durchaus auf Papier erfolgen. Man muss argumentieren können, ob man die Ergebnisse überhaupt überblicken kann. Daher ist eine Zusammenfassung derer z.B. am Anfang oder Ende des Testberichts wichtig.
Sehr geehrter Herr Johner,
vielen Dank für Ihre Gedanken! – das klingt in der Tat interessant. Bei unseren Testautomatisierungstools liegen die Spezifikationen der Tests im Versionskontrollsystem vor, ebenso die (detaillierten) Ergebnisse.
Testergebnisse der Tests auf allen Ebenen fassen wir im Testbericht zusammen, inklusive Bewertung der Ergebnisse.
Sehr geehrter Herr Prof. Johner,
Sie schrieben in Ihrem Artikel:
„Die FDA verzichtet bei einigen Systemen darauf, dass Sie „part 11 compliant“ erstellt werden:
•Alt-Systeme, die vor dem 20. August 1997 in Betrieb waren
•Systeme, die Papierausdrucke erzeugen.
Der 21 CFR part 11 ist also nur dann anzuwenden, wenn elektronische Aufzeichnungen das Papier ersetzen.“
Dazu hätte ich eine kleine Präzisierung: bzgl. der sog. „Legacy Systeme“, also der Systeme die vor dem 20.08.97, dem Inkrafttreten des Part 11, bestanden, gelten ja einige Einschränkungen. Bspw. die Systeme mussten den damals und heute bestehenden predicate rules entsprechen und man muss besonderes Augenmerk auf Changes legen, die seit dem ggf. durchgeführt wurde.
Zum Thema „Papierausdrucke“ möchte ich sagen, dass es auch noch und dass in vielen Fällen, sog. „hybrid-Systeme“ gibt. Diese fallen natürlich auch unter den part 11,oder nicht?
Eine Frage: wenn ich die Möglichkeit habe Papierausdrucke zu machen, kann ich dann die elektronischen Daten löschen und das Papier als GMP-relevante Aufzeichnung definieren?
MfG, Chr. Kunath
Sehr geehrter Herr Kunath,
wenn Sie mit den Papieren die notwendigen Nachweise führen können, benötigen Sie die elektronischen Aufzeichnungen nicht. Gerade bei großen Datenmengen ist ein manuelles Durchsuchen und Auswerten aber oft schwierig bis unmöglich. Dann wäre Papier kein Ersatz.
Beste Grüße, Christian Johner
Hallo Herr Johner,
vielen Dank für den interessanten Beitrag. Sie haben das Thema wirklich gut zusammengefasst.
Eine Frage hab ich noch. Haben Sie noch ein paar Tips für die praktische Umsetzung des 21 CFR part 11?
Liebe Grüße,
Philipp Ott
Sehr geehrter Herr Ott,
danke für Ihre freundliche Rückmeldung. Darüber freue ich mich.
Mit der Bitte um Tipps tue ich mir noch schwer. Das hängt einfach sehr von der Art der Daten, den Werkzeugen, der IT-Landschaft und den geforderten Rollenkonzepten sowie den Dokumenten ab, die unter den part 11 fallen. Mir mehr Informationen kann ich spezifischer werden.
Viele Grüße, Christian Johner
sehr geehrter Herr Johner
ich gebe hier eine ergänzende Bemerkung zum Abschnitt „Welche Systeme und Unterlagen sind betroffen?“
Zusätzlich sind auch anwendungsspezifische Methoden und Abläufe betroffen, welche erst beim Anwender mit der Applikations-SW programmiert werden, sowie die sich aus diesen Methoden erzeugten Aufzeichnungen.
Hier sind auf Seite des Betreibers organisatorische Massnahmen nötig, dass die elektronisch erzeugten Methoden und deren Aufzeichnungen den Anforderungen nach Part 11 genügen.
Sehr geehrter Herr Johner
ich habe nun schon bei diversen Audits zu hören bekommen (speziell von US-Auditoren), dass sämtliche Dokumente mit handschriftlichen Hinweisen, Einträgen, usw. die final in PDF-Form mittels SwissID unterzeichnet werden, immer noch in Papierform archiviert werden müssen.
Erklärung: SwissID ist eine staatlich geprüfte und mittels Reisepass, von den Behörden legitimierte, elektronische Unterschriftsform (wird teilweise auch von unseren deutschen Partnern verwendet).
Die Forderung, elektronisch signierte Dokumente in Papierform archivieren zu müssen, verletzt im hohen Grad mein Logik-Verständnis, denn alle Dokumente müssen vor Unterschrift in ein PDF-file umgewandelt werden. Nach vollzogener Unterschrift ist jegliche Manipulation am PDF-file unmöglich, da eine Veränderung oder ein erneutes Einscannen die Signatur ungültig werden lässt.
Mir ist bewusst, das ein Grossteil der Anwender in Ländern arbeiten, dessen elektronische Werkzeuge zur Sicherung von Daten nicht ausreichend genug ist, aber müssen sich deshalb „sichere Systeme“ diesen unterordnen?
Vielen Dank für eine kurze Stellungnahme und freundliche Grüsse,
Norbert Köppel
Eine Forderung nach einer Speicherung auf Papier gibt es nicht. Das wäre angesichts des 21 CFR part 11 auch absurd.
Sehr geehrter Herr Johner
Ich habe ein System (PLS), in dem selbst über die Bedienung keine kritischen Entscheidungen getroffen werden (Protokolle werden dort auch nicht erzeugt).
Es laufen aber kritische Daten (für spätere Entscheidungen) auf und werden im Umlaufarchiv zwischengespeichert und später an ein übergeordnetes System weitergegeben. Gehe ich recht in der Annahme, dass das alleine ausreicht, um 21 CFR P11 für das PLS einzufordern? Ich würde dann damit den Nachweis erbringen, dass die Datenbank der Umlaufarchivs „dicht“ und integer ist.
Wenn die Daten sofort, ohne Zwischenspeicherung weitergegeben würden, sehe ich
erst einmal keinen Bedarf an 21 CFR P11 im PLS. Die Schnittstelle würde ich im Rahmen der Funktionstests überprüfen.
Vielen Dank für eine Stellungsnahme
Sehr geehrter Herr Rumpf, jedes computerisierte System, das in einem QM-Prozess eingesetzt wird, ist erst einmal validierungspflichtig.
Wenn ein System Daten nur weiterleitet, müsste man nur prüfen, ob es das auch richtig tut. Das sollte eine Sache weniger Stunden sein, das zu dokumentieren.
Wenn das System Daten auch speichert, wäre auch diese Funktionalität zu validieren.
In Ihrem System scheint es „nur“ um elektronische Dokumente zu gehen, nicht um elektronische Unterschriften. Das vereinfacht die Validierung zusätzlich.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
vielen Dank für die ausführliche Beantwortung so vieler spannender Fragen.
Ich hätte noch eine Frage zur Systemdokumentation. 21 CFR part 11 fordert von der Systemdokumentation „Kontrollen über die Verteilung, den Zugang und die Benutzung“ sowie „Versions- und Änderungskontrollverfahren“.
Nicht ganz klar ist mir jedoch, das eine Systemdokumentation im Sinne des 21 CFR part 11 genau ist und beinhalten muss. In welcher Form hat eine Systemdokumentation vorzuliegen? Als SOP oder eher als elektronisch geführte Liste?
Viele Grüße,
M. Krause
Sehr geehrter Herr Kraus,
ich würde die Systemdokumentation (z.B. Beschreibung des Systems, der Anforderungen, der Konfiguration, der Architektur, der Laufzeitumgebung usw.) von den Verfahrensbeschreibungen trennen bzw. unterscheiden. Letzte sind in der Tat als SOPs zu verstehen, die regeln, wer, wann, was tun muss, um Inputs in Outputs zu überführen.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
wenn man die Papierreduktion in der Firma vorantreiben möchte, wäre part 11 sicherlich hilfreich, jedoch die damit verbundenen Auflagen und entsprechende Risiken sind bei einem Audit auch in Betracht zu ziehen.
Wie ist folgender Vorschlag bei der FDA einzuschätzen, der bei der ISO 13485 für Entwicklungsdokumente (z.B. Dokumentation der Phasen Planning bis Design Transfer) möglich ist:
For documentation of development documents it is sufficient to print the cover page (for signatures) including the version history. The complete word document is saved as PDF version. The archive finally consists of the signed paper documents (cover including version history), the scanned signed paper documents (cover including version history) and the complete document as PDF file.
Vielen Dank für Ihre Stellungnahme.
Sehr geehrte Frau Müller,
danke für die spannende Frage. Ich bin nicht sicher, ob diese ohne weitere Informationen beantwortbar ist.
Ob damit „Compliance“ vorliegt, hängt davon ab, ob das unterschriebene Cover und der Rest des Dokuments (PDF) eindeutig miteinander verbunden sind und Fälschungen zumindest sehr erschwert sind. Das könnte z.B. dadurch gelingen, dass das eingescannte Cover-Sheet und das PDF gleichzeitig ins Archiv eingestellt wird. In diesem Fall würde der (unveränderbare) gemeinsame Zeitstempel beide digitale Dokumente untrennbar miteinander verbindet.
Noch eleganter wäre es, wenn das Archiv in der Lage ist, die Freigabe von Dokumenten direkt darin zu erteilen. Damit wäre es ein Dokumentenmanagementsystem. Über einige organisatorische Maßnahmen können sogar (kostenfreie) Versionsverwaltungssysteme solche Aufgaben übernehmen.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
haben Sie vielen Dank für all Ihre aufschlussreichen Informationen.
Sie haben aufgeführt, dass „wenn Sie mit den Papieren die notwendigen Nachweise führen können, benötigen Sie die elektronischen Aufzeichnungen nicht“.
Nun ist es doch für nahezu jedes Dokument (z. B. PDF-Ausdruck) möglich dieses über diverse Tools nachträglich oder vor dem Druck zu manipulieren. Mir ist daher noch nicht ganz klar, warum es gestattet ist, die elektronischen Originaldaten zu löschen/überschreiben.
Muss vor dem Löschen/Überschreiben der Daten die Übereinstimmung zwischen elektronischen Daten und Papierdokument geprüft und mittels Unterschrift bestätigt werden?
Haben Sie vielen Dank
Mit freundlichen Grüßen
J. Sommer
Lieber Herr Sommer,
Vielen Dank für Ihre spannende Frage. Diese lässt sich nicht ganz pauschal mit ja oder nein beantworten. Zunächst: Die allgemeine Aussage bzgl. des Löschens steht so konkret im FDA Guidance Dokument zu Part 11:
„As long as predicate rule requirements are fully satisfied and the content and meaning of the records are preserved and archived, you can delete the electronic version of the records.“
Das Guidance Dokument nennt als Bedingung die Einhaltung der Predicate Rules bzgl. Freigabe dieses Löschvorgangs und lässt den Nachweis der Einhaltung offen. Der 21 CFR Part 11 und der EU GMP Annex 11 (letzterer ist nicht verbindlich für die Medizintechnik!) verlangen beide, dass ein menschenlesbarer Ausdruck der regulatorisch relevanten Aufzeichnung gemacht werden kann. Diesen Aspekt würden wir bei der initialen Validierung eines Systems nachweisen, auch hinsichtlich Korrektheit.
Werfen wir zusätzlich noch einen Blick in das Guidance Dokument der MHRA zu Datenintegrität, wird uns gesagt, dass wir die Archivierung von elektronischen Aufzeichnungen in Papierform machen dürfen und dabei zeigen sollen, dass wir die Rohdaten, Metadaten und weitere relevante Information zum Computerisierten System beim Archivieren erhalten. D.h. somit, dass risikobasiert entschieden werden sollte, ob ggf. eine zusätzliche Validierung für Archivierungsprozesse nötig ist, sofern das nicht bereits mit der initialen Validierung eines Systems gezeigt worden ist.
Als weitere Kontrolle könnten Sie die – eigentlich – üblichen periodischen Prüfungen von Aufzeichnungen einsetzen. Das verhindert nicht ggf. den Verlust von Daten, lässt es aber nach einer bestimmten Zeit erkennen. Falls Sie Fälschungen oder Fehler durch Unachtsamkeiten von Seite Ihrer Mitarbeiter befürchten und Sie Ihre Systeme nicht entsprechend härten können, sollten Sie zusätzlich organisatorische Massnahmen in Betracht ziehen, um die Integrität Ihrer Daten zu gewährleisten und entsprechend Zugriffe auf Systeme und Verzeichnisse einschränken oder zusätzliche Abgleiche vor dem Löschen vorsehen. Jede einzelne Löschung mit Freigabe durchzuführen halte ich in Anbetracht der Massnahmen Validierung oder organisatorischen Abläufen allerdings weder für angemessen, noch praktikabel.
Beste Grüsse, Urs Müller
Ich würde gerne mehr verstehen, wann genau 21 CFR 11 angewendet werden muss. Wir sind ein Lieferant von Kalibrierdienstleistungen, die von FDA-betroffenen Firmen bei uns einkaufen.
Nach meinem ersten Eindruck unterliegen WIR als Lieferant von „beschriebenem Papier“ nicht diesen Anforderungen. Können Sie das bestätigen?
Vielen Dank!
Sehr geehrter Herr Müller-Schöll
Im Guidance Dokument „Part 11, Electronic Records; Electronic Signatures – Scope and Application“ aus dem Jahr 2003 legt die FDA dar, was für sie der Anwendungsbereich von Part 11 ist. Explizit steht im Guidance Dokument:
„We understand that there is some confusion about the scope of part 11. Some have understood the scope of part 11 to be very broad. (…) As a result, we want to clarify that the Agency intends to interpret the scope of part 11 narrowly.“
Die FDA definiert anschliessend, dass der Part 11 für „electronic records“ gilt, die unter die Regulierungen der FDA fallen und für solche Records, die zwar ausgedruckt werden, die eigentlichen regulatorischen Arbeiten aber mit den „electronic records“ gemacht werden. Elektronische Unterschriften fallen natürlich auch unter den Part 11. Werden die elektronisch erzeugten Daten ausgedruckt und anschliessend auch via Papier weiterverwendet, fallen diese Aufzeichnungen nicht unter den Part 11. Auch an die FDA elektronisch übermittelte Unterlagen fallen unter den Part 11.
Ohne jetzt die genauen Konstellationen bzgl. Ihrer Kundenbeziehungen zu kennen, wage ich festzulegen, es liegt in der Verantwortung Ihrer Kunden, wie diese die von Ihnen erhaltenen Kalibrierberichte archivieren und ob diese dann bei der jeweiligen Kundenorganisation zu einem „electronic record“ werden.
Beste Grüsse, Urs Müller
Sehr geehrter Herr Müller
Herzlichen Dank für die schnelle und kompetente Antwort! Ich lese, dass Sie grundsätzlich bestätigen, dass wir als nicht der FDA unterliegender Lieferant zunächst nicht direkt betroffen sind. Daraus, dass wir einen FDA-Betrieb beliefern, lässt sich nicht ableiten, dass wir auch (sämtliche!!) Anforderungen der FDA erfüllen müssen. Es liegt also im Ermessen des FDA-betroffenen einkaufenden Betriebes , welche Forderungen er an uns stellt. Hier ist es wohl denkbar, dass er zur Erfüllung seiner eigenen FDA-Anforderungen solche Forderungen an uns weitergibt (z.B. solche von digitalen Signaturen). Dies geschieht aber auf vertraglicher Basis und nicht weil WIR den FDA-Anforderungen unterliegen würden.
Beste Grüsse C. Müller-Schöll
Frage, gibt es schon eine Bewertung wie sich die Änderungen im 21 CFR 820 auf Part11 auswirken werden? https://www.johner-institut.de/blog/qualitaetsmanagement-iso-13485/21-cfr-820/
Stand heute werden ja die im z.b. 820 genannten Records und Signaturen als für Part11 relevat herangezogen (DHF, Batch Recors etc.) mit den verweis auf ISO13485 gelten dann nur noch die dort geliteten Records?
Vielen Dank.
Sehr geehrter Herr Kewitz,
danke für die wichtige Frage!
Es ist wie Sie vermuten: Wenn Sie regulatorisch relevante Dokumente und Nachweise / Aufzeichnungen elektronisch unterschreiben wollen, greift der Part 11. Daran ändert der Verweis im Part 820 auf die ISO 13485 nichts, welcher große Teile des Parts 820 ersetzen wird.
Mit den besten Grüßen
Christian Johner
Sehr geehrter Herr Johner,
vielen Dank für Ihre ausführliche Erklärungen. Ich hätte eine Frage bzgl. den grundsätzlichen Anforderungen an die elektronischen Signatur gem. 21 CFR part 11: den Namen des Unterzeichners, Datum und Uhrzeit der Unterschrift und was mit der Unterschrift bestätigt wird (Autorenschaft, Prüfung, Freigabe). Bei uns verwendete elektronische Signatur inkludiert nicht den Grund für die Unterschrift. Ist somit unsere elektronische Signatur nicht mehr FDA-konform? Wenn ich es richtig verstanden haben, können alternativ Dokumente handschriftlich unterschrieben, eingescannt und eingereicht werden und somit die Anforderungen von part 11 umgehen. Ist dies korrekt? Können einzelne Dokumente aus dem Einreichungspaket, die elektronisch erstellt, aber papierbasiert von allen Beteiligten per wet-ink unterschrieben werden als eingescannte Kopien zur Einreichung verwendet werden?
Vielen Dank und beste Grüße
Maria Wicker
Liebe Frau Wicker,
Die Anforderung des 21 CFR Part 11 scheint in dieser Hinsicht ziemlich eindeutig zu sein: es braucht neben Name und Datum eine dritte Informationskomponente, FDA nennt diese „Meaning“. Wenn diese fehlt, haben Sie keine 21 CFR Part 11 konforme elektronische Unterschrift. Der Part 11 verlangt, dass diese Information auch bei einem Ausdruck visualisiert werden muss.
In Einreichungsunterlagen finden sich grundsätzlich immer händisch unterschriebene, eingescannte Unterlagen, z.B. Prüfberichte von Laboren. Meine Kollegen gehen ebenfalls den Weg, die zu unterschreibenden Unterlagen händisch signieren zu lassen und anschliessend den Stapel an einzureichenden Unterlagen in PDF-Form damit zu ergänzen.
Bzgl. Einreichung hat die FDA Ihre Wege entsprechend beschrieben: https://www.fda.gov/medical-devices/premarket-notification-510k/510k-submission-process. In kürze wird für bestimmte Einreichungen nur noch das eSTAR Verfahren verfügbar sein, mit eSTAR spielen Unterschriften keine Rolle mehr.
Freundliche Grüsse
Urs Müller