
Die DAkkS, die Deutsche Akkreditierungsstelle, ist die nationale Akkreditierungsbehörde der Bundesrepublik Deutschland.
1. Die Aufgaben der DAkkS
Die Aufgabe der DAkkS besteht in erster Linie darin, die Sicherheit, wissenschaftliche Reproduzierbarkeit und Vergleichbarkeit von sog. Konformitätsbestätigungen zu gewährleisten. Dazu überwacht sie die Konformitätsbewertungsstellen im staatlichen Akkreditierungsverfahren. Beispiele für Konformitätsbestätigungen sind:
- Labormessungen
- Zertifikate von Zertifizierungsstellen
- Kalibrierscheine
Die DAkkS beurteilt die fachliche Kompetenz, die wissenschaftliche Tauglichkeit der Prüfmethoden und die Unparteilichkeit der Stellen.
Durch internationale Abkommen sorgt die DAkkS zudem dafür, dass akkreditierte Zertifikate oder Prüfberichte auch außerhalb Deutschlands anerkannt werden, um den Handel zu vereinfachen.
2. Die Zuständigkeiten der DAkkS
a) Keine Zuständigkeit für Benannte Stellen für Medizinprodukte
Im Bereich des Medizinprodukterechts hat Deutschland (ähnlich Frankreich und andere Länder) entschieden, bei der Qualitätssicherung durch Benannte Stellen für die MDR und IVDR nicht auf die Akkreditierung durch die DAkkS (oder deren Pendants) zu setzen, sondern auf eine Benennung durch eine andere Behörde, im Falle Deutschland durch die ZLG.
Für den Bereich Konformitätsbewertung von Medizinprodukten wird die DAkkS deshalb nicht tätig. In Deutschland ist die ZLG die alleinig zuständige Behörde für die Benannten Stellen und deren Unterauftragnehmer, einschließlich Laboratorien (vgl. §§ 17–22 MPDG).
Eine Benannte Stelle ist gemäß ISO/IEC 17065 organisiert. Sie ist sowohl berechtigt als auch verpflichtet, alle Anforderungen der MDR, insbesondere des Artikels 10 der MDR, zu überprüfen. Das umfasst auch das Qualitätsmanagementsystem nach ISO 13485.
b) Zuständigkeit für Zertifizierungsstellen für Managementsysteme (ISO 13485)
Hersteller oder Zulieferer können sich freiwillig und unabhängig von der Konformitätsbewertung durch eine Benannte Stelle zertifizieren lassen, die auch eine nach ISO/IEC 17021-1 akkreditierte Zertifizierungsstelle ist, nach EN ISO 13485. Ein solches Zertifikat könnte helfen, die Prozesse zur Einhaltung der regulatorischen Anforderungen an die Organisation (Hersteller, Inverkehrbringer, Zulieferer) nachzuweisen, etwa die Anforderungen aus Artikel 10 der MDR.
Zertifizierungsstellen, die keine Benannten Stellen sind, aber Zertifikate nach EN ISO 13485 ausstellen wollen, müssen selbst nach ISO/IEC 17021-1 akkreditiert sein. Für diese Akkreditierung ist die DAkkS zuständig.
Diese Akkreditierung durch die DAkkS darf nicht mit der Benennung durch die ZLG verwechselt werden.
Interessanterweise werden die Benannten Stellen auch in ihrer Rolle als Zertifizierungsstelle von der ZLG im Auftrag der DAkkS auditiert. Die eigentliche Akkreditierung spricht aber die DAkkS aus, die Benennung erfolgt durch die ZLG.
Es gibt Organisationen, die „nur“ Zertifizierungsstellen sind, und Prüforganisationen, die sowohl „Zertifizierungsstelle“ als auch „Benannte Stelle“ sind.
c) Weitere Zuständigkeiten
Die DAkkS ist neben den Zertifizierungsstellen u. a. auch für die Prüfstellen/Labore und die Kalibrierstellen zuständig.
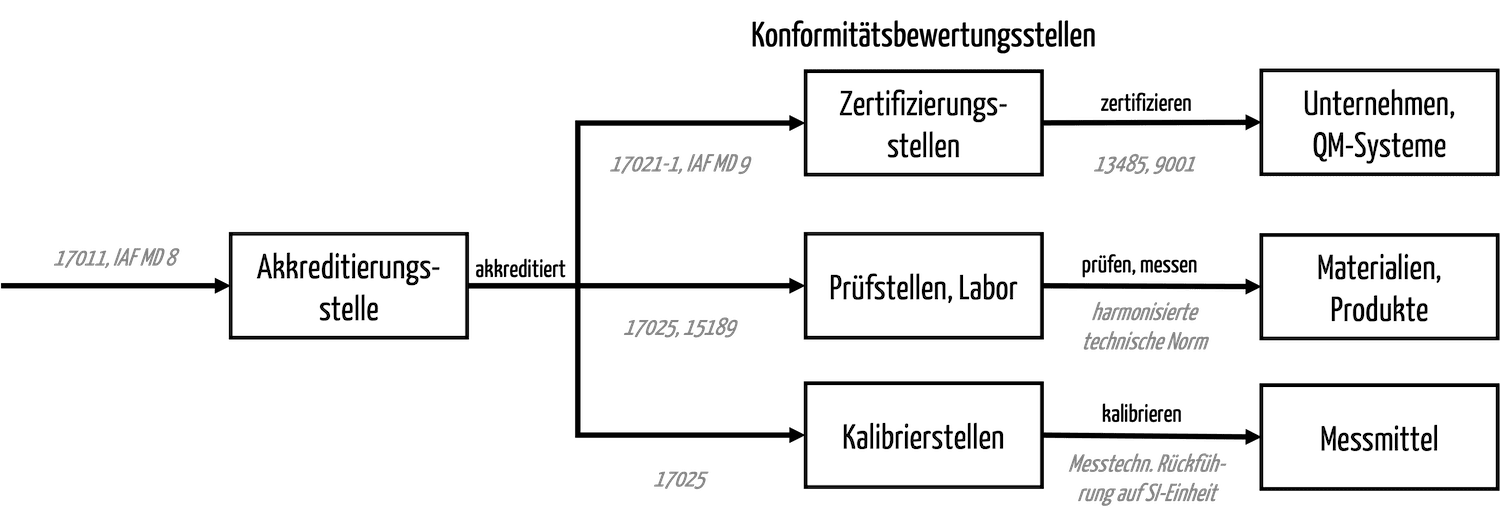
d) DAkkS-Zertifizierung – gibt es das?
Aus dem oben geschriebenen folgt, dass es keine DAkkS-Zertifizierung gibt, sondern Akkreditierungen. Beispielsweise akkreditiert die DAkkS die medizinischen Labore nach ISO 15189.
Es ist die Aufgabe der von der DAkkS akkreditierten Zertifizierungsstellen (z. B. für Managementsysteme), die Zertifizierungen vorzunehmen und die Zertifikate auszustellen.
Das Johner Institut unterstützt Hersteller von Medizinprodukten und IVD, Zertifizierungen durch Zertifizierstellen zu erlangen, beispielsweise Zertifizierungen nach ISO 13485.
Das Johner Institut unterstützt auch medizinische Labore bei deren Akkreditierung durch die DAkkS.
3. DAkkS: Regulatorische Einordnung
a) Rechtlicher Rahmen
Die EU-Akkreditierungsstellen unterliegen selbst den regulatorischen Anforderungen der Verordnung (EG) 765/2008. Die EN ISO/IEC 17011 konkretisiert diese gesetzlichen Anforderungen.
Alle EU-Akkreditierungsstellen unterziehen sich regelmäßig einer Peer-Evaluation gemäß Art. 10 der oben genannten Verordnung. Die EA (European co-operation for Accreditation) organisiert diese Evaluationen. Sie stellt sicher, dass die nationalen Akkreditierungsstellen harmonisiert arbeiten, soweit das Unionsrecht dies verlangt.
Die DAkkS veröffentlicht die Ergebnisse dieser Evaluation, hier z. B. die Ergebnisse aus dem Jahr 2022.
b) Gegenseitige Anerkennung
Innerhalb der EU/EWR folgt die gegenseitige Anerkennung der DAkkS-Akkreditierung und der Konformitätsbestätigungen von DAkkS-akkreditierten Laboren und Zertifizierungsstellen aus Art. 11 Abs. 2 VO (EG) 765/2008. Damit gelten solche Zertifikate im gesamten EU-Binnenmarkt und im EWR.
Soll eine gegenseitige Anerkennung mit Drittstaaten, aber innerhalb der WTO-Staaten erfolgen, muss das in völkerrechtlichen Gegenseitigkeitsabkommen der EU oder in bilateralen Verträgen mit Deutschland vereinbart sein. Diese Abkommen müssen explizit auf die MLA/MRA-Ebene des International Accreditation Forums (IAF) oder der International Laboratory Accreditation (ILAC) verweisen. Dies ist im Bereich Medizinprodukte und Managementsystem zu ISO 13485 nicht der Fall (vgl. FAQ 62 Medical Device Single Audit Program (MDSAP) und siehe unten Frage 9).
c) Sonderfall Managementsysteme nach ISO 13485
Regulatorischer Rahmen
Der Bereich der Zertifizierung von Managementsystemen nach ISO 13458 ist freiwillig. Es bestehen keine rechtlich zwingenden Bindungen zur gegenseitigen Anerkennung über das IAF-MLA. Die DAkkS hat aber die Anforderungen von IAF zur ISO 13485 für den Bereich der freiwilligen Zertifizierung in ihre Verwaltungspraxis übernommen.
Für diesen Bereich gelten insbesondere die Dokumente IAF MD 8 für die Konkretisierung der ISO/IEC 17011 und IAF MD:9 für die Konkretisierung der Anforderungen der ISO/IEC 17021-1.
Diese sollen die Akkreditierungsstellen mindestens einfordern. Die Dokumente haben allerdings keine Außenwirkung. Es sind weder Normen noch Gesetze. Weder Hersteller noch Konformitätsbewertungsstellen können sich direkt darauf berufen oder sind daran gebunden. Es handelt sich vielmehr um Verwaltungsvorschriften, die die Akkreditierungsstellen als Mitglieder dieser Organisation ggf. in ihre Verwaltungspraxis übernehmen.
Die DAkkS führt eine Liste der übernommenen internationalen Regeln.
Auswirkungen
Ob die Mindestanforderungen von IAF-Regeln, EA-Regeln oder ILAC-Regeln eingehalten werden, spielt nur eine Rolle im Rahmen der Peer-Evaluation zwischen den Akkreditierungsstellen. Werden Mindestanforderungen unterschritten, könnte dies bei der Peer-Evaluation zu Problemen führen. Erst in der konkreten Situation muss durch das Evaluierungsteam geklärt werden, ob die Abweichung die Mindestanforderungen erfüllt oder nicht.
Es ist deshalb zulässig, dass in bestimmten Regionen strengere Anforderungen gelten und eingehalten werden müssen, als bei IAF oder ILAC definiert.
4. Aufregung um die Akkreditierungspraxis (obsolet)
Im Laufe des Jahres 2024 machten Hersteller, Verbände und Zertifizierungsstellen Stimmung gegen die DAkkS. Sie wurde als Beispiel geschmäht, wie die deutsche Bürokratie jede Innovation im Keim erstickt.
Inzwischen hat die DAkkS ihre Praxis geändert, sodass dieser Abschnitt nicht mehr relevant ist. Wegen der FAQ ist er weiterhin online verfügbar.
Es wurden Vorwürfe erhoben, dass dieser Abschnitt zu einseitig pro DAkkS sei.
Organisationen wollten ihre öffentlich geäußerte Kritik nicht mehr zitiert sehen. Das Thema scheint so vermint zu sein, dass nur noch wenige Akteure öffentlich Stellung beziehen wollten.
Das Johner Institut war und ist darum bemüht, alle Fakten ausgewogen und korrekt darzustellen. Weil ihm bisher keine Hinweise auf sachliche Fehler gemeldet wurden, hat es beschlossen, den Abschnitt mit diesem Hinweis zu versehen und weiter online verfügbar zu lassen.
Frage 1: Was löste die Empörung über die DAkkS aus?
Auf den Urkunden nach ISO/IEC 17021-1 für Stellen, die nach EN ISO 13485 zertifizieren, nimmt die DAkkS seit Juli 2023 eine explizite Beschreibung zum „regulativen Geltungsbereich“ auf.
Es wird über einen Alleingang der DAkkS geklagt und befürchtet, dass bereits ausgestellte und gültige Zertifikate an Anerkennung verlieren und die Hersteller mit einer deutschen Zertifizierungsstelle benachteiligt würden. Solch eine Befürchtung äußert auch Medtech Online in einem Beitrag vom Februar 2024.
Frage 2: Was hat die DAkkS tatsächlich geändert?
Die Urkunden wurden geändert …
Die DAkkS hat seit Juli 2023 die Akkreditierungsurkunden neu gestaltet. Bisher sahen sie aus wie in Abb. 2 gezeigt.

Aus weiter unten genannten Gründen hat die DAkkS einen Klarstellungsbedarf gesehen. Sie ergänzt jetzt unter dem Zusatz „wie nachfolgend bezeichnet“ eine ausführliche Erläuterung des Geltungsbereichs zu den „anwendbaren regulatorischen“ Anforderungen, die durch den Anhang zur EN ISO 13485 bestätigt werden sollen (s. Abb. 3).

Neu ist also eine Erläuterung im Kapitel A1., was der Zusatz „EN“ bei „EN ISO 13485“ bedeuten soll.
Es ist also tatsächlich so, dass die DAkkS die Darstellung geändert hat und den Geltungsbereich ausführlicher beschreibt.
Die konkrete Akkreditierungsbestätigung bezieht sich aber immer noch auf dieselbe harmonisierte Norm, nämlich die EN ISO 13485 in der jeweils geltenden harmonisierten Fassung. Die Bestätigung wurde für das geltende Unionsrecht erteilt und nicht außerhalb des Unionsrechts.
Die DAkkS hat ausweislich der Datenbank der akkreditierten Stellen (in der die Akkreditierungsurkunden abrufbar sind) schon immer nur Akkreditierungen für die „DIN EN ISO 13485“ erteilt. Es lässt sich keine Akkreditierungsurkunde finden, in der eine ISO 13485 ohne den Zusatz „EN“ akkreditiert worden wäre.
Die DAkkS bestätigt, dass sie nur Akkreditierungen für den EU/EWR-Rechtsbereich erteilt habe, was durch die Angabe „EN ISO 13485“ ausgedrückt würde. Denn nur die EN-Norm enthalte die regulativen Anforderungen zur ISO 13485:2016 in der EU.
… aber die Aussage bleibt gleich
Nach Aussage der DAkkS bestätigen die alten wie die neuen Urkunden genau dasselbe: Eine nach ISO/IEC 17021 akkreditierte Zertifizierungsstelle kann Qualitätsmanagementsysteme nach EN ISO 13485 zertifizieren. Also Qualitätsmanagementsysteme, die die in den Anhängen der EN-Version der ISO 13485 genannten regulatorischen Anforderungen an die Prozesse berücksichtigen. Genau das ist das Ziel der europäischen Harmonisierung dieser Norm.
Im Gegensatz zu den Herstellern und Verbänden kann die DAkkS deshalb keine geänderte Praxis erkennen. Sie spricht vielmehr von einer Klarstellung.
Eine Akkreditierung nach DIN EN ISO 13485 bedeutet nicht, dass der Rechtsbereich nur auf Deutschland beschränkt ist. Auch ein Hersteller, der in der Schweiz oder in Indien sitzt und in die EU liefern will, kann zertifiziert werden für die Anforderungen gemäß der EN ISO 13485.
Die Abkürzung DIN steht nur für das deutsche Vorwort und die autorisierte deutsche Übersetzung der Norm „EN ISO 13485“, also einschließlich der EU-Anhänge.
Die Abkürzung EN bedeutet: Die Zertifizierungsstelle kann kompetent beurteilen, ob durch das QM-System des Herstellers die regulativen Anforderungen aus den Anhängen ZA1 bis ZA3 der ISO 13485, die auf die regulatorischen Anforderungen der MDR bzw. IVD einzahlen, umgesetzt wurden.
Frage 3: Weshalb hat die DAkkS diese Änderung vorgenommen?
Anlass war die Beschwerde einer für die Zulassung von Medizinprodukten zuständigen nationalen Behörde im Mittleren Osten. Sie beschwerte sich bei der DAkkS darüber, dass ein Hersteller seine Produkte in diesem Land in den Verkehr gebracht hatte, ohne über Grundkenntnisse sowie die erforderlichen Prozesse und Meldewege des dort geltenden Medizinprodukterechts zu verfügen.
Dieser Hersteller verfügte über ein zertifiziertes QM-System nach „EN ISO 13485“ unter einer deutschen DAkkS-Akkreditierung und legte dieses Zertifikat (unerlaubt) als Nachweis seiner Kompetenz vor.
Die nachfolgenden Auseinandersetzungen haben nach Einschätzung der DAkkS gezeigt, dass ein Klarstellungsbedarf besteht zur Bedeutung der Angabe „EN“ vor der Nennung der Norm ISO 13485 auf den Akkreditierungsurkunden.
Frage 4: Wie begründet die DAkkS den „regulativen Geltungsbereich“ bei den Akkreditierungen?
Die ISO 13485:2016 unterscheidet sich in ihrer Konformitätserklärung von anderen Managementsystemnormen nach ISO/IEC 17021 (wie ISO 9001). Während andere Managementsysteme nur die Konformität mit der Managementsystemnorm bestätigen, verlangt die ISO 13485:2016 explizit, dass die Zertifizierung auch die Konformität der Prozesse beim zertifizierten Kunden zur Umsetzung der geltenden gesetzlichen Anforderungen umfasst.
Dies ergibt sich aus mehr als 20 Fundstellen in der Norm ISO 13485. Beispielhaft können folgende Textstellen herangezogen werden:
„In verschiedenen Zuständigkeitsbereichen gelten regulatorische Anforderungen an die Anwendung von Qualitätsmanagementsystemen durch Organisationen mit unterschiedlichen Rollen in der Lieferkette für Medizinprodukte. Aus diesem Grund erwartet diese internationale Norm von der Organisation, dass
– sie ihre Rolle(n) unter anwendbaren regulatorischen Anforderungen identifiziert;
– sie die regulatorischen Anforderungen identifiziert, die für ihre Tätigkeiten unter diesen Rollen gelten;
– sie diese anwendbaren regulatorischen Anforderungen in ihr Qualitätsmanagementsystem einbindet.Die Begriffsbestimmungen in den anwendbaren regulatorischen Anforderungen unterscheiden sich von Land zu Land und von Region zu Region. Die Organisation muss verstehen, wie die Begriffsbestimmungen in dieser internationalen Norm angesichts regulatorischer Begriffsbestimmungen in den Zuständigkeitsbereichen, in denen die Medizinprodukte verfügbar gemacht werden, zu interpretieren sind.“
ISO 13485, Kap. 01 „Allgemeines“
Eine Zertifizierung muss die Übereinstimmung mit folgenden Anforderungen sicherstellen:
„Die Organisation muss entsprechend den Anforderungen dieser Internationalen Norm und den anwendbaren regulatorischen Anforderungen ein Qualitätsmanagementsystem dokumentieren und dessen Wirksamkeit aufrechterhalten.
Die Organisation muss jede Anforderung, jedes Verfahren, jede Tätigkeit oder Regelung erstellen, implementieren und aufrechterhalten, die nach dieser internationalen Norm oder anwendbaren regulatorischen Anforderungen zu dokumentieren sind.
Die Organisation muss die Rolle(n) dokumentieren, die von der Organisation unter den anwendbaren regulatorischen Anforderungen übernommen wird/werden.“
Tz. 4.1.1 ISO 13485
„Die Organisation muss: (…) e) Aufzeichnungen erstellen und aufrechterhalten, die erforderlich sind, um die Übereinstimmung mit dieser internationalen Norm und den anwendbaren regulatorischen Anforderungen darzulegen“
Tz. 4.1.2 ISO 13485
„Die Organisation muss diese Qualitätsmanagementsystemprozesse in Übereinstimmung mit den Anforderungen dieser internationalen Norm und den anwendbaren regulatorischen Anforderungen leiten und lenken“
„An diesen Prozessen vorzunehmende Änderungen müsse: (…) c) in Übereinstimmung mit den Anforderungen dieser Internationalen Norm und den anwendbaren regulatorischen Anforderungen gelenkt werden.“
Tz. 4.1.4 ISO 13485
„Die Organisation muss ihre Verantwortung zur Konformität mit dieser internationalen Norm und Kundenforderungen sowie anwendbaren regulatorischen Anforderungen für ausgegliederte Prozesse behalten“
Tz. 4.1.5 ISO 13485
Wenn folglich eine Zertifizierungsstelle eine Organisation nach „EN ISO 13485“ zertifizieren will, muss sie beurteilen können, ob die Prozesse der ISO 13485 und den ggf. anwendbaren regulatorischen Anforderungen entsprechen, die für die Inverkehrbringung beim konkreten Hersteller anzuwenden sind und die zertifiziert werden sollen.
Dazu muss die Zertifizierungsstelle Personal beschäftigen, das Kenntnisse über die anwendbaren regulatorischen Anforderungen besitzt. Nur so kann sie beurteilen, ob das Qualitätsmanagementsystem des zu zertifizierenden Kunden die regulatorischen Anforderungen an ihn, den Hersteller/Zulieferer (nicht an das Produkt!) und an die dazu notwendigen Prozesse richtig umsetzt.
Frage 5: Schränkt die DAkkS damit den Geltungsbereich auf die EU ein?
Kurze Version der Antwort
Nein. Die akkreditierten Zertifizierungsstellen können Kunden mit Sitz auf der ganzen Welt bedienen und diesen durch die Zertifizierung bestätigen, dass sie die Anforderungen der EN ISO 13485 und damit die regulativen Prozesse der MDR und IVDR gemäß den Anhängen ZA durch ihr Qualitätsmanagementsystem erfüllen.
Wünschen die Zertifizierungsstellen (für ihre Kunden) einen breiteren regulatorischen Geltungsbereich als die EU/EWR, so müssen sie ihre Kompetenz nachweisen (d. h. Schulung der Auditoren) und diesen Geltungsbereich beantragen.
Benannte Stellen sind, wie oben dargestellt, nicht betroffen. Diese können in allen Rechtsbereichen zertifizieren, in denen sie benannt sind, einschließlich des Qualitätsmanagementsystems. Hersteller, die mit von der ZLG benannten Stellen arbeiten oder mit z. B. nach MDSAP benannten Stellen, sind also nicht betroffen.
Längere Version der Antwort
Zwar hat die DAkkS bisher die Akkreditierungen an Zertifizierungsstellen nur zur EN ISO 13485 erteilt. Es ist aber auch für deutsche Zertifizierungsstellen gemäß ISO/IEC 17021 möglich, im Rahmen des Akkreditierungsverfahrens nachzuweisen, dass sie Personal beschäftigen, das sich auch mit regulativen Systemen außerhalb der MDR/IVD auskennt, z. B. denen der „MDSAP-Staaten“ oder Taiwans.
In anderen Worten: Falls eine Zertifizierungsstelle nach ISO/IEC 17021 die eben genannten Voraussetzungen erfüllt, kann ihr die DAkkS im Akkreditierungsverfahren die Kompetenz in anderen regulativen Bereichen außerhalb der MDR/IVDR bestätigen.
Entweder gibt es für einige Staaten eigene Normen-Anhänge zur ISO 13485 (z. B. für Kanada: CAN/CSA ISO 13485:2016), oder es werden andere regulative Anforderungen an die QM-Systeme oder Prozesse oder Gegenseitigkeitsabkommen direkt beantragt und bestätigt (z. B. für die USA: 21 CFR Part 820 – Quality System Regulation; 21 CFR Part 803, 21 CFR Part 806, 21 CFR Part 807 – Subparts A-D; 21 CFR Part 821 – Device Tracking) (soweit anwendbar).
Ein uneingeschränkter bzw. nicht spezifizierter „globaler“ Geltungsbereich ist aus den o. g. Gründen nicht möglich. Denn eine Akkreditierung bestätigt das Vorliegen einer konkreten Kompetenz der Zertifizierungsstelle.
Frage 6: Macht die DAkkS einen Alleingang?
Bezüglich der Kompetenz des Personals in Zertifizierungsstellen
Die DAkkS verneint das. Sie hat die weltweiten Mindestanforderungen des International Accreditation Forum (IAF) zur Norm ISO 13485 in ihre Verwaltungspraxis übernommen (s. o.), damit sich die unter einer DAkkS-Akkreditierung zertifizierten Hersteller/Zulieferer auf das IAF-MLA zu ISO 13485 berufen können. Die DAkkS bestätigt deshalb den Zertifizierungsstellen auf den Akkreditierungsurkunden ausdrücklich, dass diese die Anforderungen der ISO/IEC 17021-1 erfüllen und auch die zusätzlichen internationalen Mindestanforderungen gemäß IAF MD 9.
Das IAF fordert im Mandatory Document (MD) 9 explizit, dass die „Accreditation Bodies“ (z. B. die DAkkS) überprüfen, dass die CAB (Conformity Assessment Bodies), also die Zertifizierungsstellen, über die notwendigen Kenntnisse des anwendbaren Rechtsrahmens verfügen (s. Zeile 2 Tabelle B2 in Abb. 4).
Das betrifft bei den CAB sowohl die Auditoren als auch Personen, die die Auditergebnisse prüfen und über die Ausstellung der Zertifikate entscheiden.
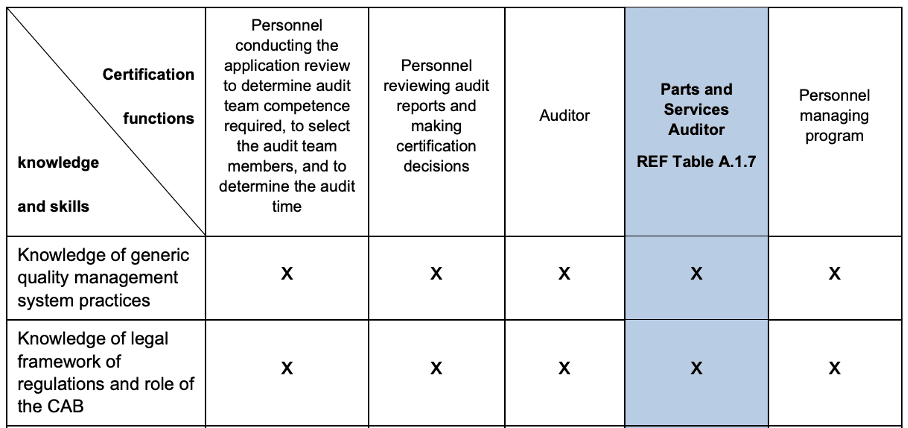
Daraus lässt sich schließen, dass weltweit alle Akkreditierungsstellen unter dem IAF-MLA verpflichtet sind zu überprüfen, ob die akkreditierte Zertifizierungsstelle auch über die Kenntnisse der „anwendbaren regulatorischen Anforderungen“ im Sinne der ISO 13485 verfügt, die die Zertifizierungsstelle ausdrücklich beantragt hat.
Bezüglich der Urkunde
Die DAkkS sieht auch hier keinen Alleingang. Die VO (EG) 765/2008 verpflichte alle EU-Akkreditierungsstellen dazu, mindestens die Anforderungen der ISO/IEC 17011 einzuhalten. Dazu zählt die Angabe des Geltungsbereichs.
„Der Geltungsbereich der Akkreditierung muss mindestens folgende Angaben enthalten. Für Zertifizierungsstellen:
die Normen, normativen Dokumente und/oder die gesetzlichen Anforderungen, nach denen Managementsysteme, Produkte, Prozesse und Dienstleistungen oder Personen zertifiziert werden, wo zutreffend.“
7.8.3 ISO/IEC 17011
Dieser europäischen Pflichtangabe komme die DAkkS durch den Verweis auf die europäisch harmonisierte Norm „EN“ ISO 13485 nach, die in deren Anhängen ZA „die gesetzlichen Anforderungen“ genannt werden.
In keinem Wirtschaftsbereich sind die Akkreditierungsurkunden zwischen den Akkreditierungsstellen in der konkreten Darstellung exakt abgestimmt. Das ist u. a. aufgrund der Vielfalt der Technik und der unterschiedlichen Anforderungen des jeweiligen nationalen Verwaltungsrechts, das nicht europäisch oder international harmonisiert ist, nur schwer möglich. Allerdings sind durch die Regelungen in der ISO/IEC 17011 sowie für den Bereich ISO 13485 durch das IAF MD 8 Mindestangaben definiert worden, die alle Akkreditierungsstellen wiedergeben müssen. Speziell: Alle Akkreditierungsstellen setzen das „EN“ oder ein anderes Kürzel vor die ISO 13485, um den regulativen Geltungsbereich zu beschreiben.
Frage 7: Was sind die Auswirkungen auf die Medizinproduktehersteller?
Der Geltungsbereich auf einer Akkreditierungsurkunde der DAkkS zu ISO/IEC 17021 hat zunächst nichts mit den Herstellern zu tun. Es geht allein um die Frage, welche Kompetenz einer Zertifizierungsstelle nach ISO/IEC 17021 durch die DAkkS bestätigt wird.
Auswirkungen auf die Inverkehrbringung in Europa
Die Inverkehrbringung von Medizinprodukten setzt (abhängig von der Klasse dieser Produkte) voraus, dass eine Benannte Stelle einbezogen wird. Benannte Stellen unterliegen der Aufsicht der ZLG und nicht der DAkkS.
Eine Auswirkung auf die Inverkehrbringung von Medizinprodukten in Europa gibt es deshalb nicht. Alle Hersteller, die mit einer von der ZLG benannten Stelle arbeiten, sind nicht betroffen. An der Benennung durch die ZLG ändert sich nichts.
Auswirkung auf die Inverkehrbringung in anderen Ländern
Es gibt keine unmittelbaren Auswirkungen auf die Inverkehrbringung von Produkten in anderen Ländern, weil die Zertifizierung nach ISO 13485 durch eine Zertifizierungsstelle nach ISO/IEC 17021 weder ein zwingender noch ein hinreichender Nachweis für die Inverkehrbringung von Medizinprodukten ist.
Allerdings kann es für Hersteller sinnvoll sein, eine Zertifizierung nach ISO 13485 in Verbindung mit anwendbaren regulatorischen Anforderungen nachzuweisen, wenn deshalb eine Benannte Stelle oder eine Medical Device Authority ihr Prüfprogramm reduziert. Das könnte z. B. in den Ländern interessant sein, die am MDSAP-Programm teilnehmen.
Auch hier gilt, dass eine Benannte Stelle, die am MDSAP-Programm teilnimmt, nicht betroffen ist von einer Änderung der Akkreditierungsurkunde nach ISO/IEC 17021. Die am MDSAP teilnehmenden Staaten können die Ergebnisse zum QM-System nach ISO 13485 mit den zusätzlichen regulatorischen Anforderungen berücksichtigen.
Unabhängig von der Art der Zertifizierung (entweder durch eine Benannte Stelle oder eine Zertifizierungsstelle nach ISO/IEC 17021) müssen und mussten die Hersteller schon immer die rechtlichen Anforderungen der jeweiligen Länder ermitteln und einhalten, in denen sie die Verantwortung für die Inverkehrbringung übernehmen wollen.
Andernfalls müssen sie dieses Risiko und die gesetzlich geforderten Aktivitäten vertraglich auf einen (lokalen) Inverkehrbringer bzw. Käufer übertragen. In diesem Fall muss das QM-System nur den Kundenannahmeprozess beschreiben und steuern, sodass eine Wirksamkeit der Verträge sichergestellt ist.
Auswirkung auf die Audits und Zertifikate von Zertifizierungsstellen nach ISO/IEC 17021
Die Akkreditierungsurkunde betrifft das Rechtsverhältnis zwischen der DAkkS und der jeweiligen Zertifizierungsstelle.
Ob einem Hersteller ein Zertifikat entzogen wird, entscheidet somit nicht die DAkkS, sondern allein die Zertifizierungsstelle anhand der Verträge zwischen Hersteller und Zertifizierungsstelle.
Für Hersteller/Inverkehrbringer gibt es derzeit auch keine Auswirkungen auf schon nach EN ISO 13485 erteilte Zertifikate von Zertifizierungsstellen nach ISO/IEC 17021 (die keine Benannten Stellen sind). Denn nach Aussage der DAkkS hat sich keine Änderung der Akkreditierung ergeben. Die alten Urkunden wurden für die „EN“ ISO 13485 ausgestellt, genauso wie die neuen Urkunden, die „EN“ ISO 13485 bestätigen.
Ein Grund, Zertifikate einzuschränken oder zurückzuziehen, ist nicht ersichtlich und nach Auskunft der DAkkS bisher nicht aufgetreten.
Mehr zu den Benannten Stellen weiter unten.
Die Zertifizierungsstellen verpflichten sich typischerweise vertraglich gegenüber den Herstellern, eine akkreditierte Zertifizierung zur EN ISO 13485 durchzuführen. Falls die Zertifizierungsstelle feststellen sollte, dass ein Zertifikat noch nicht vom Geltungsbereich der Akkreditierungsurkunde abgedeckt ist, muss sie einen Erweiterungsantrag bei der DAkkS stellen. Andernfalls läuft sie Gefahr, ihre vertraglichen Pflichten gegenüber dem Hersteller nicht erfüllen zu können.
Die Zertifizierungsstelle hat typischerweise kein Recht, den Vertrag mit dem zertifizierten Kunden zu kündigen, weil sie die zugesagte Akkreditierung nicht besitzt. Es kann jederzeit vorkommen, dass die DAkkS Nichtkonformitäten bei einer Zertifizierungsstelle feststellt. Dann ist die Zertifizierungsstelle in der Pflicht, durch Korrekturmaßnahmen die Akkreditierungsvoraussetzung wiederherzustellen und dem Hersteller nachzuweisen.
Hersteller müssen somit bezüglich ihrer Zertifikate nichts tun.
Frage 8: Was sind die Auswirkungen auf die Zulieferer?
Die Aussage zu den Herstellern gilt analog auch für die Zulieferer. Das heißt: Auch die Zulieferer müssen nichts tun. Hingegen ist deren akkreditierte Zertifizierungsstelle oder Benannte Stelle in der vertraglichen Pflicht, alles Notwendige und Zumutbare einzuleiten, um die Leistung wie vertraglich vereinbart unter einer gütigen Akkreditierung oder einer gültigen Benennung zu erbringen.
Frage 9: Was sind die Auswirkungen auf die Benannten Stellen?
Keine Auswirkungen auf die Tätigkeit Benannter Stellen in der EU
Hersteller bzw. Inverkehrbringer, die die Bewertung ihres QM-Systems nach ISO 13485 von einer Benannten Stelle aus Deutschland oder der EU vornehmen lassen, sind von der beschriebenen Änderung der Akkreditierungsurkunde nicht betroffen.
Benannte Stellen, die in der EU nach ISO/IEC 17065 organisiert sind und von einer „Medical Device Authority“ wie der ZLG benannt wurden, dürfen mehrere sog. „Evaluierungstätigkeiten“ erbringen, z. B.:
- Prüfung der Produkte oder Baumuster
- Bewertung von QM-Systemen oder die Produktionskontrolle
- Zertifizierung des Produkts auf diesen Grundlagen, woraufhin der Hersteller das Produkt in Verkehr bringen darf
Das hier diskutierte Problem der Zertifizierungsstellen nach ISO/IEC 17021 hat keinen Bezug zu Bestätigungen durch Zertifizierungsstellen nach ISO/IEC 17065 oder durch von einer „Medical Device Authority“ Benannten Stelle. Dies liegt daran, dass es sich entweder um eine andere Akkreditierung oder gar keine Akkreditierung, sondern um eine Benennung handelt.
Daraus folgt: Selbst wenn ein Prüfunternehmen sowohl eine Benennung durch die ZLG und ggf. eine weitere ausländische „Medical Device Authority“ hat und zusätzlich in einem weiteren Geschäftsbereich auch als Zertifizierungsstelle nach ISO/IEC 17021 tätig ist, handelt es sich dennoch um völlig getrennte Rechtsbereiche. Die Tätigkeit als Benannte Stelle ist nicht von der Akkreditierung nach ISO/IEC 17021 betroffen und umgekehrt.
Keine Auswirkungen auf die Tätigkeit Benannter Stellen außerhalb der EU
Die Hersteller müssen die regulatorischen Anforderungen der Länder einhalten. In den Ländern, die am MDSAP-Programm teilnehmen, weisen Hersteller die Konformität mit der ISO 13485 und den zusätzlichen lokalen, regulativen Anforderungen des Staates nach, in dem sie Medizinprodukte in Verkehr bringen wollen. Um Doppelprüfungen zu vermeiden, wird dies im Hinblick auf die Konformitätsaussage zur ISO 13485 und die MDSAP-Kriterien, einschließlich der regulativen Anforderungen, von anderen MDSAP-Staaten akzeptiert. Die EU nimmt nicht an MDSAP teil.
Benannte Stellen, die Medizinprodukte einschl. der QM-Systeme dieser Hersteller prüfen, sind — anders als Zertifizierungsstellen, die nur Zertifikate nach ISO/IEC 13485 für die QM-Systeme erteilen — nicht nach ISO/IEC 17021, sondern nach ISO/IEC 17065 akkreditiert oder für z. B. MDSAP „benannt“. Gemäß dem Dokument „IMDRF MDSAP WG N3“ und FAQ Nr. 62 besteht keine Verbindung zwischen einer Akkreditierung nach ISO/IEC 17021 gemäß dem IAF-MLA und dem MDSAP-Programm. Die Akkreditierung Benannter Stellen nach ISO/IEC 17021 durch die DAkkS ist deshalb nicht relevant für die Zulassung nach „MDSAP AS P0034“.
Die „MDSAP AS P0034“ spricht auch nur über die Einbeziehung von „European Notified Bodies“, also Benannten Stellen, und gerade nicht von akkreditierten Zertifizierungsstellen nach ISO/IEC 17021. (Quellen: FDA, IMDRF)
Einige Prüfunternehmen, die in der EU als „Benannte Stelle“ tätig sind, haben sich zusätzlich als „MDSAP Auditing Organization“ benennen lassen. Sie dürfen auch „MDSAP-Zertifikate“ ausstellen, die die Bewertung enthalten, ob der Hersteller auch die QM-Anforderungen dieser Länder erfüllt.
Hersteller, denen die Konformität ihres Produkts mit der ISO 13485 und mit den Anforderungen aus dem teilnehmenden MDSAP-Staat attestiert wurde, haben den Nachweis umfassend für alle MDSAP-Staaten erbracht.
Frage 10: Was sind die Auswirkungen auf die Zertifizierungsstellen?
Die DAkkS erwartet von den Zertifizierungsstellen nach ISO/IEC 17021 die im IAF MD 9 geforderten Kompetenzen. Daraus folgt:
Falls die Zertifizierungsstellen von der DAkkS einen breiteren Geltungsbereich als EU/EWR wünschen, müssen sie die Kompetenzen ihrer Auditoren durch entsprechende Schulungsnachweise für die beantragten Geltungsbereiche belegen. Sie müssen der Behörde Evidenz vorlegen, dass die regulatorischen Anforderungen bei den Auditoren bekannt und verstanden sind; denn nur dann können diese beurteilen, ob das QM-System die Prozesse wirksam implementiert hat.
Dazu können die Zertifizierungsstellen beispielsweise Schulungen für die Teilnahme der Auditoren am MDSAP-Programm für MDSAP-Länder nutzen. Sind die Unterschiede im regulativen System im Vergleich zur EU gering, können sogar interne Schulungen der Stelle genügen (z. B. für die Schweiz).
Entsprechende Erweiterungsanträge können bei der DAkkS gestellt werden.
Sollte eine Zertifizierungsstelle nach ISO/IEC 17021 Handlungsbedarf beim Geltungsbereich feststellen und daraufhin einen Erweiterungsantrag stellen, besteht keine Notwendigkeit weiterer behördlicher Maßnahmen. Insbesondere entzieht die DAkkS keine Zertifikate (s. o.).
Alle deutschen Stellen dürfen nur bei der DAkkS akkreditiert werden. Es gilt das „Sitzlandprinzip“.
Betroffen könnten Zertifizierungsstellen sein, welche bisher nur nach ISO/IEC 17021 für den Geltungsbereich „DIN EN ISO 13485“ akkreditiert waren, aber dennoch Zertifikate erteilt haben, die nicht lediglich die Konformität zur „DIN EN ISO 13485“ bestätigt haben. Der DAkkS ist solch ein Fall bisher nicht bekannt.
Zusammenfassung
i) Die Situation wurde sehr unterschiedlich beurteilt
Die Beurteilung der Situation fiel durch die Hersteller, Zertifizierungsstellen und Benannten Stellen einerseits und die DAkkS andererseits unterschiedlich aus:
- Hersteller fühlten sich bereits genug gegängelt durch MDR und IVDR, durch Benannte Stellen und durch die EU (mit ihren MDCG-Dokumenten und der schleppenden Harmonisierung). Jetzt noch die DAkkS. Das empfanden viele als Zumutung. Daher instrumentalisierten die Hersteller Verbände, Ministerien und Normungsorganisationen. Ein regelrechter Shitstorm braute sich zusammen.
- Die DAkkS konnte das nicht nachvollziehen, weil sie nur eine Klarstellung, aber keine Änderung vorgenommen habe.
ii) Nicht alle, die jammern, sind betroffen
Selbst wenn man die Klarstellung der DAkkS auf ihren Akkreditierungsurkunden zur Abkürzung „EN“ als Änderung interpretierte, waren nicht alle Organisationen betroffen, die dagegen Sturm liefen:
- Die Akkreditierungsurkunden betreffen nur das Rechtsverhältnis zwischen der DAkkS und den Zertifizierungsstellen nach ISO/IEC 17021. Nicht betroffen sind Benannte Stellen und Hersteller.
- Hersteller mussten schon immer „nur“ die rechtlichen Anforderungen jener Länder ermitteln, einhalten und dies nachweisen, in denen sie ihre Produkte in den Verkehr bringen. Hier kann sich also keine Änderung aus der Zertifizierung des QM-Systems ergeben.
iii) Was konnten wir aus der Sache lernen?
Vielleicht ließ sich aus dieser (unnötig?) anstrengenden Situation zumindest etwas lernen:
- Redet miteinander
Eine fachlich fundierte Kommunikation miteinander und nicht übereinander hilft, negative Emotionen, Eskalationen und Rechtsstreite zu verhindern oder zumindest einzudämmen. Das erfordert zeitnahe und verbindliche Antworten auf Anfragen. - Nutzt den „Risk-based Approach“
Der Nutzen regulatorischer Anforderungen für die Sicherheit und die Versorgung der Patienten skaliert nicht mit der Anzahl der Anforderungen. Der Aufwand, um diese Anforderungen zu erfüllen, schon. Daher sollte auch hier der „Risk-based Approach“ gelten.
Wenn ein Hersteller nicht weiß, wie er in einem Land schnell und wirksam unsichere Medizinprodukte zurückrufen kann und muss, dann stellt das ein hohes Risiko dar. Daher ist es sinnvoll, dass Hersteller (auch) die Vigilanzanforderungen der jeweiligen Länder kennen und erfüllen müssen, sowie dass die Zertifizierungsstellen oder die Benannten Stellen das auch prüfen (können). - Vergesst die Patienten nicht
Wir beobachten längst, dass die Hersteller deshalb nicht mehr im gleichen Maß bezahlbare Medizinprodukte anbieten, um möglichst alle Patienten zu diagnostizieren und zu therapieren. Das ist für uns alle ein Problem. Wir alle sind potenzielle Patienten. Beispielsweise hat ein sehr großer Hersteller sein Portfolio halbiert. Entfernt wurden nicht nur ältere Produkte, sondern auch Produkte für Kinder und für Patienten mit seltenen Krankheiten.
Anders formuliert: Selbst, wenn am Ende alle Recht behalten, ist niemandem geholfen. - Vereinfacht das System
Das regulatorische System ist so kompliziert geworden, dass es dieses weiteren umfangreichen Artikels bedurfte, um nur einen Aspekt zu beleuchten.
Wir haben mit den oben dargestellten Informationen und FAQ versucht, mehr Licht ins Dunkel zu bringen. Wir haben dazu die DAkkS um Stellungnahme zu Einzelfragen gebeten und Antworten erhalten, die wir entsprechend berücksichtigt haben.
In Summe erscheint das Problem aus Sicht der Hersteller weniger problematisch zu sein, als es sich zunächst vermuten ließ. Benannte Stellen sind nicht betroffen und Zertifizierungsstellen nach ISO/IEC 17021 nur in bestimmten Auslandskonstellationen außerhalb der EU.
Änderungshistorie
- 2025-01-27: Kapitel 2.b) leicht überarbeitet und um Details ergänzt
- 2025-01-05: Der Streit um die Akkreditierungspraxis ist inzwischen beigelegt. Daher ist dieser Themenkomplex obsolet und aus Gründen der Dokumentation im vierten Abschnitt zusammengefasst. Abschnitt 2.d) eingefügt, der klarmacht, dass es keine DAkkS-Zertifizierungen gibt.
- 2024-03-25: Öffentliche, aber zurückgezogene Zitate und Referenzen auf speziellen Wunsch der zitierten Personen entfernt. Hinweis zu Beginn eingefügt. Link auf Beitrag von Medtech Online ergänzt.
- 2024-01-16: Erste Version des Artikels veröffentlicht



Die Texte in die dunkel (schwarz?) markiert sind sind leider nur sehe sehr schwer oder gar nicht lesbar. Könne Sie da eine andere Markerfarbe verwenden?
Lieber Herr Fiedler,
vielen Dank für Ihren wichtigen Hinweis!
Wir haben keine Markierungen im Artikel beabsichtigt und nach einer möglichen Ursache gesucht.
Wir hoffen, das Problem nun behoben zu haben. Geben Sie uns gerne nochmal Bescheid, falls dem nicht so sein sollte (am besten mit einem Screenshot per E-Mail an info@johner-institut.de).
Ich wünsche Ihnen einen schönen Tag!
Herzliche Grüße
Tea Bodrusic
Sehr geehrter Herr Prof. Johner,
herzlichen Dank für den Artikel und ihre vortreffliche Analyse. Es sind eben oftmals internationale Vorgaben, die innerhalb Deutschlands von der DAkkS umgesetzt werden müssen. Würden wir die internationale Anerkennung verlieren, wäre dies für alle in Deutschland ansässigen Beteiligten sicherlich ein weitaus größeres Problem!
Als langjährige DAkkS-Mitarbeiterin bis Januar 2023 habe ich einiges von dem „Shitstorm“ gegen die DAkkS mit abbekommen, der ja oftmals auch auf dem Rücken der Verfahrensmanager mit Widerspruchsverfahren ausgetragen wird und das ganze System weiter verlangsamt.
Herzliche Grüße
Dr. Nina Rählert
Herzlichen Dank für Ihre wertschätzende Antwort, Frau Dr. Rählert!
Darüber freuen wir uns sehr. Denn es ist uns ein Anliegen, einen kleinen Beitrag zu leisten, um unnötige Shitstorms zu vermeiden. Denn diese kosten alle Seiten nur Energie, die wir an anderer Stelle besser einsetzen können.
Uns erreichen bereits die nächsten Fragen, und wir werden alles daransetzen, auch diese zu beantworten.
Mit nochmaligem Dank und mit herzlichen Grüßen, Christian Johner
Sehr geehrter Prof. Dr. Johner,
wir erleben die DAkkS als eine der langsamsten Firmen überhaupt und im Vergleich zu anderen Akkreditierungsstellen unter ILAC als Einzelkämpfer mit sehr individuellen Interpretationen zum Regelwerk.
Bearbeiter sind quasi nie erreichbar, Verfahrensmanager auch nicht, Rechnungen sind nicht nachvollziehbar (vielleicht auch weil sie erst 5 Jahre nach Leistungserbringung eintrudeln), Beschwerden werden nicht bearbeitet. Daher beglückwünsche ich Sie dazu eine Stellungnahme der DAkkS erhalten zu haben, dass bleibt uns und auch anderen Kunden ja verwehrt.
Die Kritik, die Verbände, Hersteller und Zertifizierungsstellen an der DAkkS üben, ist berechtigt und sollte zur Reflexion des Agierens führen. Denn, ja, die Arbeitsweise kostet alle Geld, bringt Patienten nichts und sorgt nur für Frust und verhärtete Fronten.
MfG
Schmidt
Danke für Ihre wertvollen Gedanken, Herr Schmidt, die ich alle völlig nachvollziehen kann.
Ähnliche Nachrichten erreichen uns oft. Und auch ich habe den Eindruck, dass in diesem regulatorischen Wahnsinn die Patienten auf der Strecke bleiben.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner!
Ich möchte Ihnen von meiner Erfahrung als QM eines österreichischen Entwicklungsdienstleisters für Medizinprodukte berichten:
Unsere deutsche benannte Stelle hat uns Anfang Dezember 2024 kontaktiert um unseren Geltungsbereich auf dem EN ISO 13485 Zertifikat „umzuformulieren“, weil laut deren wörtlicher Aussage ein „Autobus voller Auditoren“ notwendig wäre.
Das Ganze geschah vom Zeitpunkt her ca. 5 Wochen vor dem vor einem halben Jahr vereinbarten Re-Zertifizierungsaudit und unter der Drohung, dass wir sonst nicht auditiert werden könnten.
Als Begründung wurden die DAkkS-Aktivitäten und die IAF MD9 genannt.
Als Entwicklungsdienstleister brauchen wir für zukünftige Kunden einen breiten Geltungsbereich und nach zwei Wochen des Hin-und-Hers von Geltungsbereichsversionen, denke ich wir haben einen breiteren Geltungsbereich als vorher.
Das Re-Zertifizierungsaudit haben wir mittlerweile ohne Hauptabweichung bestanden.
Die ganze Thematik hat allerdings zwei Wochen „genervt“!
Darum kann ich nicht zustimmen, dass die DAkkS-Aktivitäten keine Auswirkungen auf Hersteller haben.
Mit freundlichen Grüßen.
Andreas Weinfurter