
Im QSIT (Quality System Inspection Technique) weist die FDA Ihre Inspektoren an, wie diese die Konformität von Qualitätsmanagementsystemen mit den regulatorischen Forderungen des 21 CFR part 820 prüfen sollen. Für Medizinproduktehersteller dient der QSIT damit nicht nur zur Vorbereitung auf FDA Inspektionen, sondern auch als Anregung für das Vorgehen bei eigenen internen Audits.
Ziel des QSIT
Mit dem QSIT (Quality System Inspection Technique) will die FDA den eigenen Inspektoren eine Methodik an die Hand geben, um schnell und systematisch die Konformität von Qualitätsmanagement-Systemen mit regulatorischen Anforderungen insbesondere denen des 21 CFR part 820 zu prüfen.
Ansatz des QSITs
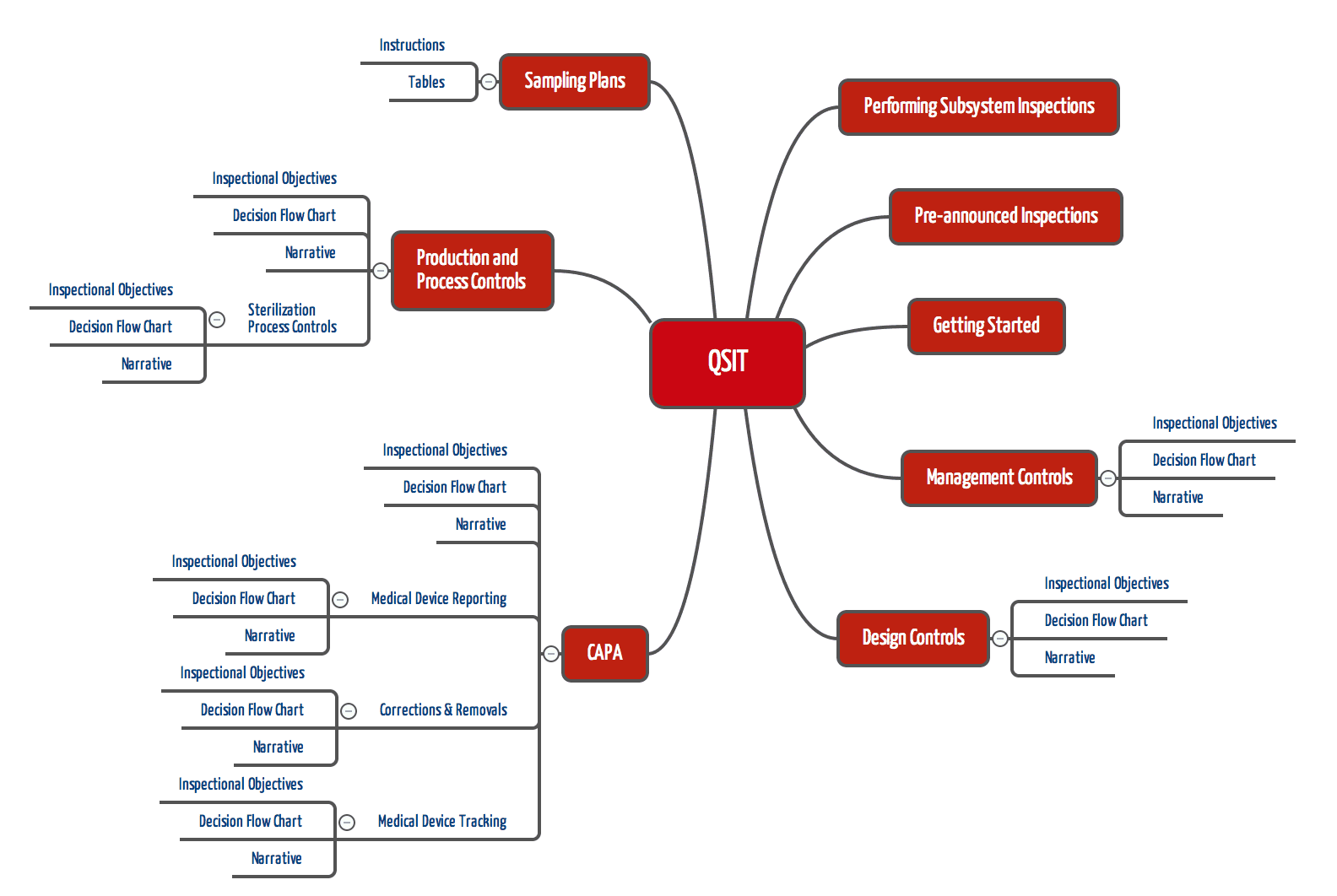
FDA-Inspektoren sollen Inspektionen nicht button-up durchführen (z.B. bei einem konkreten Problem beginnen und dann die Ursache im QM-System suchen), sondern Top-down. Dabei sollen die Inspektoren die in den folgenden Kapiteln genannten „Subsysteme“ einzeln prüfen:
- Management Controls
- Design Controls
- CAPA mit drei Sub-Subsystemen („Medical Device Reporting“, „Corrections & Removals“ sowie „Medical Device Tracking“)
- Production and Process Controls mit dem Sub-Subsystem „Sterilization Process“
Mit dieser Hierarchie gibt die FDA mit dem QSIT eine Priorität und eine Reihenfolge vor. Der Top-Down-Ansatz soll sicherstellen, dass Inspektoren in ausreichender Breite prüfen.
Für jedes dieser (Sub-)Subsysteme legt die FDA im fest
- Die Prüfkriterien („Inspectional Objectives“)
- Das Vorgehen in Form eines Ablaufdiagramms („Decision Flow Chart“) und
- Das Vorgehen in beschreibender Form („Narrative“)
Ebenfalls in diesem allgemeinen Kapitel fordert die FDA von ihren Inspektoren, dass diese mit den Verfahrensbeschreibungen starten und dann in die Aufzeichnungen vorstoßen sollen.
QSIT: Quality System Inspection Technique: Zum Vergrößern klicken
Was der QSIT im Detail fordert
Kapitel „Performing Subsystem Inspections“
Den Inhalt dieses Kapitels finden Sie bereits oben (Kapitel Ansatz des QSITs) beschrieben.
Kapitel „Preannounced Inspections“
Dieses Kapitel weißt die Inspektoren auf die Formalitäten hin und erwähnt, dass sie zwar eine Kopie des QM-Handbuchs verlangen sollen, dass die Firmen das aber nicht einreichen müssen.
Kapitel „Getting Started“
Dieses mit vier Sätzen sehr kurze Kapitel weißt die Inspektoren an, zu Beginn jeder Inspektion mit dem QM-Beauftragten zu sprechen und sich einen Überblick über das QM-System geben zu lassen.
Kapitel „Management Controls“
Nun geht es ans erste „Subsystem“, die „Management Controls“.
Ziele („Inspectional Objectives“)
Als Ziele der Prüfung nennt der QSIT beispielsweise:
- Prüfen, ob festgelegt und dokumentiert sind
- die Verfahrensanweisungen für die Qualitätspolitik, die Managementbewertung und die Audits,
- der Qualitätsplan
- die Verfahrensbeschreibungen und Arbeitsanweisungen
- Prüfen, dass die Qualitätspolitik und die Qualitätsziele umgesetzt werden.
- Prüfen, dass die Aufbauorganisation der Firma Verantwortlichkeiten und Befugnisse festlegt und dass die Firma die notwendigen Ressourcen bestimmt.
Insgesamt sieben Ziele formuliert der QSIT, nur die ersten drei finden Sie hier genannt.
Ablaufdiagramm („Decision Flow Chart“)
Das Ablaufdiagramm enthält
- Fragen (Checklisten)
- Eine Reihenfolge, in diese Fragen gestellt werden sollen
- Einen Bezug zur Nummer des jeweiligen oben genannten Prüfziels
- Eine Referenz auf den entsprechenden Paragrafen des 21 CFR part 820.
Erläuterungen („Narrative“)
Die Erläuterungen gehen nun auf jeden der sieben Punkte ein. Sie stellen auch fest:
If you found major nonconformances (as defined in the Compliance Program, Part V) in your review of the management or other subsystems that indicate management with executive responsibility is not ensuring the establishment and maintenance of an adequate quality system, you may cite this deficiency on your FDA 483. This cite should not be used routinely, but should be used in those situations where major portions of a quality system have not been established and maintained or whenever there is a total lack of a quality system.
When you have made that determination and have completed your FDA 483, or decided no FDA 483 is needed, you may proceed to your final discussion with Management, or the official closeout meeting with the firm.
Kapitel „Design Controls“
Dieses Kapitel gibt den Inspektoren in 15 konkreten Anweisungen wie zu prüfen, ob
- die Design Inputs existieren
- die Design Outputs identifiziert sind
- die Verifizierung und Validierung durchgeführt wurden
- es eine Risikoanalyse gibt,
- die Design Reviews durchgeführt wurden
- usw.
Dazu soll der Inspektor ein einzelnes Entwicklungsprojekt auswählen und bei Produkten, die Software enthalten, entscheiden, ob auch die Software-Validierung zu prüfen ist.
Das Ablaufdiagramm wiederholt diese Anweisungen nahezu unverändert, wieder mit Referenz auf die Prüfanweisungen (hier die genannten 15) und die Paragrafen des 21 CFR part 820.
Der Begriff Ablaufdiagramm würde mehr erwarten lassen: Es ist nur eine sequenzielle Abfolge von Prüfschritten ohne Fallunterscheidungen.
Kapitel „Corrective and Preventive Actions (CAPA)“
Dieses Kapitel des QSIT unterscheidet sich von den vorangegangen darin, dass es nicht nur aus dem üblichen „Dreisprung“ Prüfziele – Ablaufdiagramm – Erläuterungen besteht, sondern zusätzlich Unterkapitel enthält, die wiederum nach dem „Dreisprung“ aufgebaut sind. Diese Unterkapitel betreffen
- Die Meldung von Zwischenfällen
- Korrekturen und Rückrufe
- Nachverolgung von Medizinprodukten
Kapitel „Production and Process Controls“
Auch dieses Kapitel enthält mit „Sterilization Process“ ein Unterkapitel.
Wie die Kapitelüberschrift nahelegt, geht es hier um die Produktion und Prozesskontrolle. Wichtige Schlagworte sind die Prozessvalidierung, die „Device History Records“ und die Validierung von Prozesssoftware.
Fazit und Zusammenfassung
Der FDA QSIT (Quality System Inspection Technique) gibt eine relativ konkrete Vorgabe, welche Aspekte eines QM-Systems in welcher Reihenfolge zu prüfen sind. Dazu benennt der QSIT insgesamt 8 Subsysteme bzw. Sub-Subsysteme, jeweils mit
- Prüfzielen
- Ablaufdiagramm
- Erläuterungen
Die Prüfziele sind als knappe und konkrete Fragen bzw. Anweisungen formuliert.
Die Ablaufdiagramme wiederholen diese Ziele meist nur. Worin deren Mehrwert genau besteht (außer der zusätzlichen Referenz auf die Kapitel des 21 CFR part 820) erschließt sich nicht sofort, weil es rein sequenzielle Abläufe ohne Fallunterscheidungen sind.
Wie konkret Aspekte zu prüfen sind, beispielsweise ein Design Input, findet sich zumindest teilweise in den jeweiligen Erläuterungen.
Der QSIT ist zwar über 15 Jahre alt, was man den sicher nicht von einem Designer entworfenen Grafiken und 80er-Jahre Powerpoint-Cliparts deutlich ansieht. Dennoch sei jedem Hersteller empfohlen, diesen QSIT nicht nur zu lesen, sondern als Grundlage für interne Audits und die Vorbereitung auf FDA Inspektionen zu nutzen.



Hallo,
vielen Dank für die guten Ratschläge.
Viele Grüße,
Marko Sommer