
Die EU plant, mit der neuen EU-Verordnung zu Batterien und Altbatterien („Batterieverordnung“) die EU die bestehende Richtlinie 2006/66/EG zu ersetzen.
Dieser Artikel klärt darüber auf,
- welche Medizinproduktehersteller und welche Medizinprodukte von der Batterieverordnung betroffen sind,
- welche Anforderungen diese Verordnung stellt und
- wie die Hersteller am besten vorgehen, um diese Anforderungen zu erfüllen.
1. Hintergrund
a) Zielsetzung
Mit der Batterieverordnung möchte die EU den Rechtsrahmen für Batterien modernisieren. Die Verordnung ist eine Reaktion auf die Forderung nach einer umfassenden Überarbeitung und Erweiterung der EU-Gesetzgebung für den gesamten Lebenszyklus von Batterien.
Das Ziel besteht darin, die Ressourceneffizienz zu steigern und die zirkuläre Wertschöpfung zu fördern. Die Batterieverordnung ist ein integraler Bestandteil des European Green Deal.
b) Aktueller Stand der Batterieverordnung
Die Verordnung (EU) 2023/1542 des Europäischen Parlaments und des Rates vom 12. Juli 2023 über Batterien und Altbatterien, zur Änderung der Richtlinie 2008/98/EG und der Verordnung (EU) 2019/1020 und zur Aufhebung der Richtlinie 2006/66/EG hat die EU am 28. Juli 2023 im Amtsblatt veröffentlicht.
Download der EU-Batterieverordnung, Stand erste Lesung vom 14. Juni 2023, Deutsch
2. Anwendungsbereich der Batterieverordnung
a) Welche Produkte sind betroffen?
Der Artikel 1 der Batterieverordnung legt deren Anwendungsbereich fest.
Diese Verordnung enthält Anforderungen an die Nachhaltigkeit, Sicherheit, Kennzeichnung und Information, die das Inverkehrbringen und die Inbetriebnahme von Batterien in der Union ermöglichen. Darüber hinaus enthält sie die Mindestvorschriften für die erweiterte Herstellerverantwortung, die Sammlung und Behandlung und für die Berichterstattung.
Diese Verordnung gilt für alle Batterien, namentlich Gerätebatterien, Starterbatterien, Traktionsbatterien und Industriebatterien, unabhängig von Form, Volumen, Gewicht, Gestaltung, stofflicher Zusammensetzung, Verwendung oder Zweck. Sie gilt außerdem für Batterien, die in andere Produkte eingebaut sind oder ihnen beigefügt werden.
Dabei legt die Batterieverordnung folgende Definitionen fest.
eine Batterie, die gekapselt ist und die weniger als 5 kg wiegt.
Die Verordnung definiert:
ein Elektro- oder Elektronikgerät im Sinne der Richtlinie 2012/19/EU, das vollständig oder teilweise mit einer Batterie betrieben wird oder betrieben werden kann;
Die Richtlinie 2012/19/EU (Elektro- und Elektronik-Altgeräte) schließt gemäß Anhang I alle Medizinprodukte oder Zubehör ein, mit Ausnahme aller implantierten und infektiösen Produkte.
Die Batterieverordnung betrifft somit alle Medizinprodukte (implantierbare Produkte und Produkte, die durch die Nutzung infiziert werden, ausgenommen), in denen Gerätebatterien verbaut sind.
Eine einzige Ausnahme für Medizinprodukte gibt es in der Anforderung in Artikel 11 zur Entfernbarkeit und Austauschbarkeit von Gerätebatterien.
Beispiele für Medizinprodukte, die betroffen sind:
- Spritzenpumpe mit einer Backupbatterie
- Transportabler Patientenmonitor
- Abnehmbarer Laryngoskop-Griff mit Batterie
- Fahrbarer Therapieroboter mit einem Elektroantrieb, der von einer Transaktionsbatterie versorgt wird
- Ohrthermometer mit austauschbarern Knopfzelle
- Blutdruckmessgerät für den Heimgebrauch mit Standard AAA Zellen
Beispiele für Medizinprodukte, die nicht betroffen sind:
- Implantierbare Herzschrittmacher mit wiederaufladbarer Batterie (Vorgaben sind in der MDR enthalten.)
- Chirurgische Instrumente, die über den Klinikabfall entsorgt werden müssen
- Einmal-Laryngoskope mit Beleuchtung und fest verbauter Batterie, die über den Klinikabfall entsorgt werden muss
Stand alone Software
Bei Software-Medizinprodukten, die auf allgemeinen Mobilplattformen oder Tablets installiert und betrieben werden, ist der Hersteller der Hardware für die Einhaltung der Verordnung verantwortlich. Es kann notwendig sein, dass der Hersteller des Software Medizinprodukts Informationen zur Batteriekennzeichnung in die Gebrauchsanleitung des Medizinprodukts übernehmen muss, wenn der Hersteller die Software zusammen mit einem Gerät ausliefert.
b) Welche Organisationen sind betroffen?
Zunächst betrifft die Batterieverordnung alle Hersteller, die eine Batterie (einschließlich in Geräte oder Fahrzeuge eingebaute Batterien) erzeugen oder entwickeln oder erzeugen lassen und diese Batterie unter ihrem eigenen Namen oder unter ihrer eigenen Handelsmarke vermarkten oder zu eigenen Zwecken in Betrieb nehmen und erstmals im Hoheitsgebiet eines Mitgliedstaats gewerbsmäßig für den Handel oder die Verwendung abgibt.
Als Erzeuger, Einführer oder Händler zählen auch Hersteller!
Dass der Begriff des Herstellers weit gefasst ist, zeigt der Artikel III, Absatz 42. Demnach qualifizieren die folgenden Aktivitäten eine Organisation als „Hersteller“:
- mit Niederlassung in EU: Erzeugen oder konzipieren oder lassen erzeugen und vertreiben in der EU unter eigenem Namen oder unter der eigenen Handelsmarke
- mit Niederlassung in EU: Weiterverkaufen von anderen EU-Herstellern erzeugte Batterien, wenn der Name oder die Handelsmarke dieser anderen Hersteller nicht angegeben ist
- mit Niederlassung in EU: Erstmals gewerbsmäßig Abgabe von Batterien aus einem anderen Mitgliedstaat oder einem Drittland stammend
- mit Niederlassung in EU oder in einem Drittland: Direkter Verkauf über Fernabsatzverträge an Endnutzer
Welche Medizinproduktehersteller sind betroffen?
Medizinproduktehersteller fallen unter die erste Bedingung, wenn sie
- für ihr Medizinprodukt (z. B. einen Patienten-Transportmonitor) eine Batterie für ihre Bedürfnisse entwickeln und erzeugen lassen und zusammen mit dem Produkt unter eigenen Namen vertreiben.
- eine fertige gebrauchsfähige Batterie aus Batteriezellen für das Medizinprodukt selbst entwickeln und erzeugen.
Welche Medizinproduktehersteller sind nicht betroffen?
Hersteller, deren Geräte für Standardbatterien vorgesehen sind und bei denen keine Batterien ausgeliefert werden, sind nicht betroffen. Die Gebrauchsanleitung muss allerdings Hinweise zu dem zu verwendenden Batterietyp beinhalten. Das ist bereits eine Forderung der MDR.
3. Die wichtigsten Anforderungen der Verordnung
Die Batterieverordnung umfasst 13 Kapitel und 14 Anhänge (s. Abb. 1).
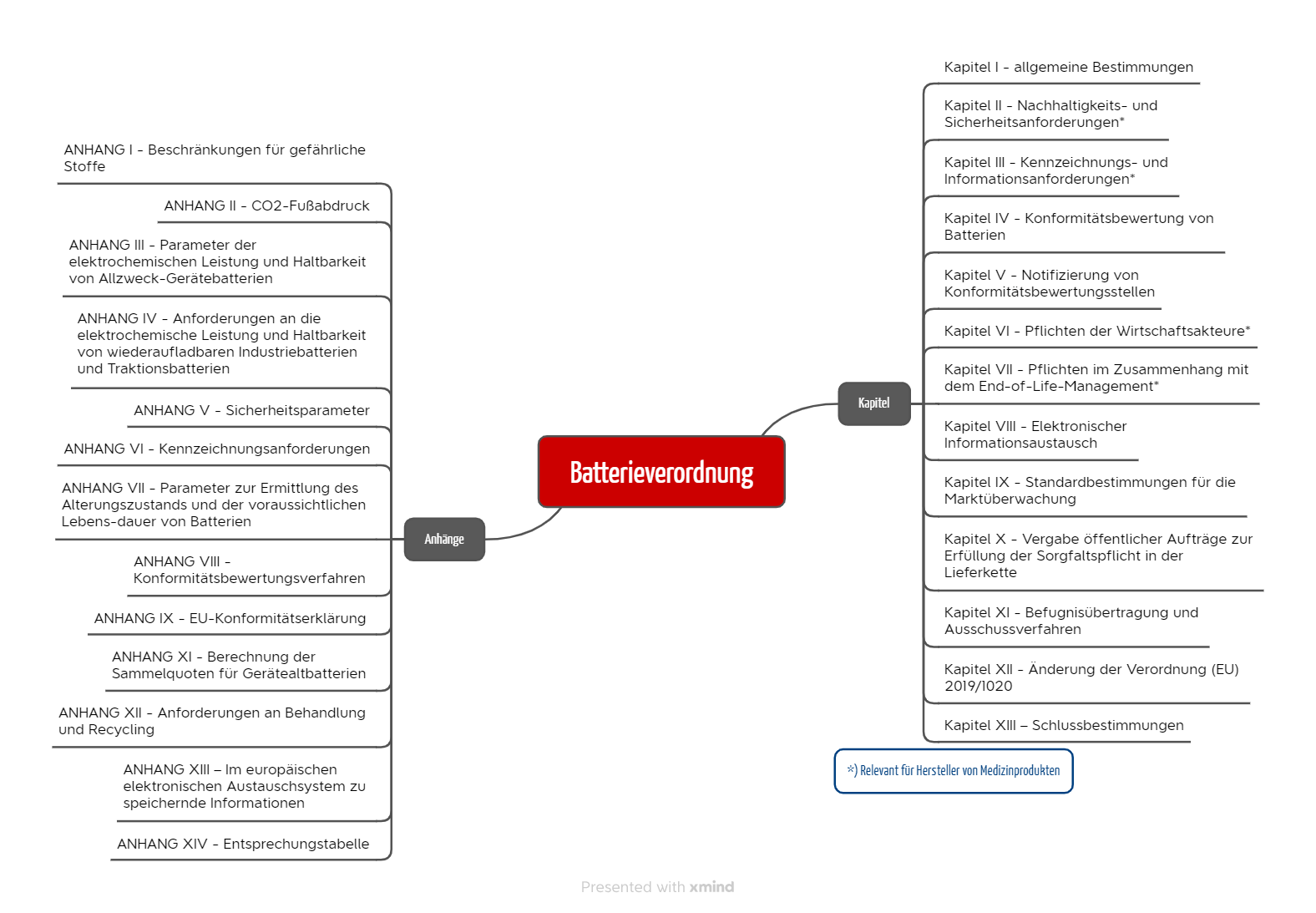
Die Anforderungen für die Hersteller von Medizinprodukten lauten:
- Erklärung zum CO2-Fußabdruck
- Entfernbarkeit und Austauschbarkeit von Gerätebatterien
- Kennzeichnungs- und Informationsanforderungen
- Elektronischer Informationsaustausch
- Pflichten der Wirtschaftsakteure
a) Erklärung zum CO2-Fußabdruck
Artikel 7 fordert, dass Hersteller von Batterien, Traktionsbatterien oder wiederaufladbaren Industriebatterien zukünftig den CO2-Fußabdruck in Form einer Erklärung abgeben müssen. Die Methoden zur Berechnung des CO2-Fußabdrucks werden von der EU vorgegeben.
Die zeitliche Einführung der Vorschriften erfolgt in drei Stufen:
- Stufe 1 – Informationspflicht zum CO2-Fußabdruck in Form einer Erklärung
- Stufe 2 – Einstufung der Batterien in Leistungsklassen
- Stufe 3 – Einführung von Höchstwerten für den CO2-Fußabdruck über den gesamten Lebensweg
Von den Anforderungen betroffen sind zunächst nur Hersteller von bestimmten Batteriearten:
- Wiederaufladbare Industriebatterien mit internem Speicher und einer Kapazität über 2 kWh
- Elektrofahrzeugbatterien
- LV-Batterien (leichte Verkehrsmittel)
Die Fristen sind für die verschiedenen Batterietypen sind geregelt in Artikel 7, Unterabschnitte 2 und 3. Die Vorgaben treffen nicht auf Gerätebatterien zu.
Medizinproduktehersteller, die Batterien erzeugen oder entwickeln oder erzeugen lassen, gelten auch als Hersteller. Sie unterliegen somit diesen Vorschriften.
b) Entfernbarkeit und Austauschbarkeit von Gerätebatterien
Die Batterieverordnung fordert, dass die Batterien entfernt und ausgetauscht werden können:
Natürliche oder juristische Personen, die Produkte, in die Gerätebatterien eingebaut sind, in Verkehr bringen, sorgen dafür, dass diese Batterien vom Endnutzer jederzeit während der Lebensdauer des Produkts leicht entfernt und ausgetauscht werden können. Diese Verpflichtung gilt nicht für einzelne Zellen oder sonstige Teile einer Batterie, sondern nur für die ganze Batterie.
Artikel 11, Absatz 1
Das bedeutet, dass Hersteller folgende Eigenschaften der Batterien sicherstellen müssen:
- Austauschbar, wenn die Lebensdauer der Batterien kürzer ist als die des Geräts
- Entfernbar spätestens am Ende der Lebensdauer
- Endbenutzer oder unabhängige Wirtschaftsakteure müssen in der Lage sein, die Batterie zu entfernen oder auszutauschen
Natürliche oder juristische Personen, die Produkte, in die Gerätebatterien eingebaut sind, in Verkehr bringen, sorgen dafür, dass den Produkten eine Betriebsanleitung und Sicherheitsinformationen für die Verwendung, das Entfernen und das Austauschen der Batterien beiliegen.
Für folgende Geräte kann das Austauschen der Batterie auf unabhängige Fachleute beschränkt bleiben:
- Abwaschbare oder abspülbare Geräte, bei denen es im Interesse der Sicherheit des Nutzers und des Geräts erforderlich ist (Bsp. Sterilisierbare Medizinprodukte)
- Geräte, bei denen die Batterie nur funktionieren kann, wenn sie in die Gerätestruktur integriert ist (z. B. gemeinsames Gehäuse, ggf. Hörgeräte)
- Professionelle medizinische Bildgebungs- und Strahlentherapiegeräte im Sinne von Artikel 2, Nr. 1 der Verordnung (EU) 2017/745 und In-vitro-Diagnostika im Sinne von Artikel 2, Nr. 2 der Verordnung (EU) 2017/746
Es ist unklar, warum die EU alle IVDR-Produkte in die Ausnahme einschließt, aber bei den Medizinprodukten nur professionelle medizinische Bildgebungs- und Strahlentherapiegeräte. Vielleicht ist es ein Fehler und es sind alle Medizinprodukte gemeint?
Wenn man die Lesart auf alle Medizinprodukte sowie IVDR-Produkte bezieht, betrifft die Anforderung der Entfernbarkeit und Austauschbarkeit die Hersteller von Medizinprodukten nicht. Dennoch sind ggf. weitere Vorgaben anwendbar.
c) Kennzeichnungs- und Informationsanforderungen
Ab dem 1. Januar 2027 sind Batterien gut sichtbar, lesbar und nicht verwischbar zu kennzeichnen. Dazu gehören die Angaben bestimmter Hauptmerkmale wie Lebensdauer, Ladekapazität, Pflicht zur getrennten Sammlung, das Vorhandensein gefährlicher Stoffe und die Sicherheitsrisiken.
Auf Batterien soll ein dauerhafter QR-Code angebracht werden, der je nach Batterietyp den Zugang ermöglichen muss zu den für die betreffende Batterie relevanten Informationen, den Batteriepass und den Informationen zum CO2-Fußabdruck.
Wiederaufladbare Industrie- und Elektrofahrzeugbatterien müssen ein Batteriemanagementsystem (BMS) enthalten, das gemäß Anhang VII die Informationen und Daten speichert, die zur Bestimmung des Alterungszustands und der voraussichtlichen Lebensdauer erforderlich sind. Diese Anforderung lässt sich bereits aus der MDR ableiten (siehe MDR Anhang I, Absatz 18.2).
Die Absätze 1 und 2 der neuen Batterieverordnung betriffen nur Erzeuger von Batterien. Absatz 3 betrifft Hersteller, die Batterietypen verwenden, für die das BMS vorgeschrieben ist und die eine Schnittstelle für die Lesbarkeit der Informationen zur Verfügung stellen müssen.
d) Elektronischer Informationsaustausch: Der Batteriepass
Für den elektronischen Informationsaustausch sollen Hersteller von LV-Batterien, Industriebatterien und Elektrofahrzeugbatterien eine elektronische Akte („Batteriepass“) erstellen. Diese Anforderungen betreffen nur Hersteller, die selbst Erzeuger einer Batterie sind. Der Batteriepass enthält Informationen über das Batteriemodell und spezifische Informationen für die einzelne Batterie gemäß Anhang XIII. Der Batteriepass wird online über einen QR-Code zugänglich gemacht.
Die im europäischen elektronischen Austauschsystem zu speichernden Informationen umfassen:
- Öffentlich zugängliche Informationen gemäß Anhang XIII, Nr. 1
- Nur notifizierten Stellen, Marktaufsichtsbehörden und der Kommission zugängliche Informationen gemäß Anhang XIII, Nr. 2 und Nr. 3
- Nur natürlichen oder juristischen Personen zugängliche Informationen gemäß Anhang XIII, Nr. 2 und Nr. 4
Die Verordnung nennt auch die Anforderungen an die Strukturierung der Informationen und an die Interoperabilität des Datenaustausches. Die Informationen basieren auf offenen Standards und sind in einem interoperablen Format dargestellt. Sie sind maschinenlesbar, strukturiert und durchsuchbar. Artikel 78 beschreibt die technische Gestaltung des Batteriepasses.
Die Anforderung betrifft nur Hersteller, die Batterien entwickeln und erzeugen und im Sinne der Verordnung als Hersteller der Batterie gelten.
f) Pflichten der Wirtschaftsakteure
In Kapitel VI der neuen Batterieverordnung werden die Pflichten der Wirtschaftsakteure festgelegt.
| Wirtschaftsakteur | Artikel | Aktivitäten Kurzfassung |
| Erzeuger | Artikel 38 | – Nachhaltigkeits- und Sicherheitsanforderungen erfüllen gemäß Kapitel II – Erstellen der technischen Unterlagen gemäß Anhang VIII – Kennzeichnung gemäß Artikel 13 – Durchführen der Konformitätsbewertung – Bei Serienprodukten stets die Konformität sicherstellen (Normen, gemeinsame Spezifikationen) – Zugang zu Werten gemäß Anhang VII im Batteriemanagementsystem ermöglichen – Korrekturmaßnahmen und Marktüberwachung – Zusammenarbeit mit nationalen Behörden |
| Händler | Artikel 42 | – Prüfen der Registrierung des Herstellers – Prüfen der CE-Kennzeichnung der Batterie – Prüfen, ob Betriebsanleitung und Sicherheitsinformationen beigelegt sind – Prüfen, ob Erzeuger und der Einführer ihre Anforderungen erfüllt haben – Bei Nichtkonformität den Erzeuger und die Marktüberwachungsbehörde unterrichten |
| Importeure, Einführer | Artikel 41 | – Vor der Inverkehrbringung die Konformität mit Artikel 6 bis 10 und 12, 13 und 14 prüfen – Vor Inverkehrbringung prüfen, ob die Batterie ein CE-Kennzeichnung trägt – Bei Nichtkonformität den Erzeuger und der Marktüberwachungsbehörde unterrichten |
| Bevollmächtigte | Artikel 40 | – Bereithaltung der EU-Konformitätserklärung – Bereithaltung des Berichts über die Überprüfung der Sorgfaltsstrategien, bestätigt durch eine notifizierte Stelle – Aushändigung aller erforderlichen Informationen und Unterlagen zum Nachweis der Konformität an die Behörde – Kooperation mit nationalen Behörden bei allen Maßnahmen zur Abwendung der Risiken |
| Zulieferer | Artikel 39 | – Den Erzeugern die zur Einhaltung der Anforderungen erforderlichen Informationen und Unterlagen zur Verfügung stellen Hinweis: Hersteller/Erzeuger müssen darauf achten, dass sie diese Informationen erhalten. |
4. Batterieverordnung und Hersteller von Medizinprodukten
a) Anwendbarkeit bei Medizinprodukteherstellern
Hersteller, die gebrauchsfertige Batterien einkaufen und verwenden
Hersteller, die gebrauchsfertige Batterien einkaufen und verwenden, müssen folgende Anforderungen beachten:
- Prüfen der Kennzeichnung
Prüfen, ob ausreichend Informationen über die beabsichtigte Verwendung der Batterie zur Verfügung stehen, damit sie ordnungsgemäß in Betrieb genommen, verwendet und am Ende ihrer Lebensdauer bewirtschaftet werden kann. Diese Anforderungen sind indirekt enthalten in der IEC 60601-1, Abschnitt 4.8 zur Verwendung von Bauteilen. - Prüfen der Konformität
Prüfen, ob die Batterie die CE-Kennzeichnung trägt und sicherstellen, dass der Händler die Informationen (Betriebsanleitung und Sicherheitsinformationen) gemäß seinen Pflichten bereitstellt. Hersteller sollten geeignete Maßnahmen ergreifen um zu gewährleisten, dass sie nur Batterien auf dem Markt bereitstellen, die mit dieser Verordnung übereinstimmen. - Bereitstellen von Informationen
Dem Endanwender Informationen zur sicheren Entsorgung und zum sicheren Austausch bereitstellen, gegebenenfalls mit den notwendigen Hilfsmitteln. - Konstruktion und Auslegung
Geräte so entwickeln, dass Batterien (sofern die Sicherheit und Leistung nicht beeinflusst wird) entfernbar und austauschbar sind; im Gerät Möglichkeiten schaffen, mit denen die Informationen aus dem Batteriemanagementsystem (BMS) angezeigt oder ausgelesen werden können
Hersteller, die Batterien entwickeln (lassen)
Hersteller, die Batterien erzeugen oder entwickeln oder erzeugen lassen und die diese Batterien unter ihrem eigenen Namen oder unter ihrer eigenen Handelsmarke vermarkten, müssen folgende Anforderungen beachten:
Diese Hersteller (Erzeuger) müssen beim Inverkehrbringen und bei der Inbetriebnahme einer Batterie (auch für eigenen Zwecke) gewährleisten, dass
- die Batterie, die in Kapitel II beschriebenen Nachhaltigkeits- und Sicherheitsanforderungen erfüllt,
- die Batterie gemäß Artikel 13 gekennzeichnet ist,
- die technischen Unterlagen gemäß Anhang VIII erstellt wurden,
- ein Konformitätsbewertungsverfahren gemäß Artikel 11 durchgeführt wurde und
- die CE-Kennzeichnung gemäß den Artikeln 19 und 20 angebracht ist.
b) Umsetzung der Batterieverordnung
Was sollten die Hersteller tun?
Die Hersteller sollten folgende Aktivitäten durchführen:
- Prüfen, ob ihre Aktivitäten in den Geltungsbereich der Verordnung fallen bzw. ob die Anforderungen an die Entfernbarkeit und Austauschbarkeit von Gerätebatterien auf die eigenen Produkte zutreffen
- Prüfen, ob für sie die Pflichten für Händler oder Importeure zutreffen und ggf. die zusätzlichen Pflichten im QM Managementsystem implementieren
- Prüfen, ob die Ausnahmen für die Entfernbarkeit und Austauschbarkeit der Batterien in Artikel 7 zutreffen. Falls nicht, müssen Hersteller die Geräte und die Batterien so konstruieren, dass die Batterien einfach entfernbar und austauschbar sind.
- Im Gerät Möglichkeiten schaffen, mit denen die Informationen aus dem Batteriemanagementsystem (BMS) angezeigt oder ausgelesen werden können.
- Implementieren der Schnittstellen und Protokolle. Es gibt unseres Wissens keinen Standard. Vom VDE gibt es ein Fact Sheet.
- Die Informationen vom Erzeuger der Batterie in die eigene Gebrauchsanleitung integrieren oder die Informationen mit beilegen.
Die Verordnung legt großen Wert auf Transparenz und Rückverfolgbarkeit, um eine umweltfreundliche Kreislaufwirtschaft zu fördern. Es kann vorkommen, dass außereuropäische Hersteller die geforderten Anforderungen, insbesondere an die Transparenz, nicht vollständig erfüllen wollen. Daher sollten Hersteller die langfristige Lieferbarkeit für Batterien frühzeitig prüfen. Gegebenenfalls sollten sie in Erwägung ziehen, einen Hersteller innerhalb der EU zu suchen (was die Absicht der EU-Kommission war).
Was sollten Hersteller vermeiden?
Die Kommission möchte die Verwendung von wiederaufladbaren und nicht wiederaufladbaren Allzweck-Gerätebatterien (Bsp. AAA Batterie) regeln, indem sie Parameter für deren elektrochemische Leistung und Haltbarkeit vorgibt.
Das Ziel besteht darin, die Anwendung von nicht wiederaufladbaren Allzweck-Gerätebatterien zu reduzieren. Die Kommission denkt sogar über ein Verbot nach.
Hersteller sollten abwägen, ob der Einsatz einer nicht wiederaufladbaren Allzweck-Gerätebatterie gegenüber einer wiederaufladbaren Batterie ökologisch gerechtfertigt ist (z. B. weil nur eingeschränktes Recycling möglich).
c) Übergangsfristen
Die Übergangsfristen betreffen nur die Erzeuger von Batterien:
| Entfernbarkeit von Batterien | Die Anforderungen an die Entfernbarkeit und Austauschbarkeit gelten ab dem 18. Februar 2027. |
| Kennzeichnung von Batterien | Allgemeine Informationen gemäß Anhang VI Teil A für alle Batterien: Artikel 13, Absatz 1: Ab dem 18. August 2026 oder 18 Monate nach dem Tag des Inkrafttretens des in Absatz 10 genannten Durchführungsrechtsakts, je nachdem, welcher Zeitpunkt der spätere ist. Symbol gemäß Anhang VI Teil B für alle Batterien: ab dem 18. August 2025 QR-Code, Anhang VI, Teil C für alle Batterien: Artikel 13, ab dem 18. Februar 2027 Für bestimmte Batteriearten sind weitere Kennzeichnungen und deren Fristen geregelt in Artikel 13, Absätze 3 bis 10. |
| Digitaler Batteriepass | Ab dem 18. Februar 2027 für LV-Batterien, Industriebatterien mit einer Kapazität von mehr als 2 kWh und Elektrofahrzeugbatterien |
5. Fazit und Zusammenfassung
Mit der Batterieverordnung möchte die EU einen Beitrag zum Umweltschutz leisten. Wenn Ihre Medizinprodukte Batterien enthalten, sollten Sie genau prüfen, ob Sie von der Verordnung betroffen sind, und die gesetzlichen Anforderungen einhalten.
Unabhängig davon können Sie einen Beitrag zum Umweltschutz leisten, indem Sie z. B. auf den Einstz von nicht wiederverwendbaren Batterien verzichten.
Das Johner Institut unterstützt Hersteller dabei herauszufinden, ob sie von der Verordnung betroffen sind, und hilft, eine regulatorische Strategie festzulegen und umzusetzen.
Melden Sie sich, falls Sie herausfinden wollen, wie diese Unterstützung erfolgen kann und wie sie Ihnen dabei hilft, Ihre Medizinprodukte schnell und gesetzeskonform in alle Märkte zu bringen.



Wie sieht’s mit BIOS Batterien aus? Wenn ein ME Geräte mit Standard-Mother-Board gesteuert wird, wird da eine BIOS-Batterie drin sein. Und wenn die entfernt wird, wird auch ggf. ein BIOS-Passwort verloren gehen und damit eine wichtige Cybersecurity Risk-Mitigation, wenn – wie beschrieben – der Austausch der Batterie nicht nur von zertifiziertem Service-Personal gemacht werden darf. Zusätzlich müsste man ja so auch erlauben, dass ME Geräte-Gehäuse geöffnet werden … von Nicht-Service-Personal…
Hätte doch letztlich zur Folge, keine Motherboards mehr verwenden zu können (wenn nicht safety & security egal).
Oder man müsste die BIOS-Batterie durch ein eigenes Produkt ersetzen. So dass Spannung bei Austausch hoch genug bleibt (um BIOS-Einstellungen zu halten). Und die Batterie „von außen zugänglich machen“. Und wenn nicht schnell genug ersetzt — dann müsste man das Gerät deaktivieren und Service zur Reaktivierung holen.
Lieber Herr Buttgereit,
vielen Dank für Ihre interessante Frage.
Ich kann Ihre Bedenken gut nachvollziehen. In meiner Erfahrung sind BIOS-Batterien auf den Motherboards bereits austauschbar. Es wäre zumindest ratsam, insbesondere wenn die Lebensdauer des Motherboards deutlich länger ist als die der Batterie. Durch diese Maßnahme möchte man verhindern, dass eine ansonsten intakte Leiterplatte möglicherweise aufgrund einer fest verbauten Batterie entsorgt werden muss. Die Verordnung erlaubt, dass der Austausch auch durch eine Fachperson durchgeführt werden kann, die nicht der Endbenutzer ist.
Die Verordnung sagt im Artikel 11, Absatz 3 weiterhin: „Die Verpflichtungen nach Absatz 1 gelten nicht, wenn eine kontinuierliche Stromversorgung erforderlich ist und eine ständige Verbindung zwischen dem Produkt und der jeweiligen Gerätebatterie erforderlich ist, um die Sicherheit des Benutzers und des Geräts zu gewährleisten oder – bei Produkten, deren Hauptfunktion die Sammlung und Bereitstellung von Daten ist – aus Gründen der Datenintegrität.“
Somit sollte das Risiko einer Sicherheitslücke weiterhin beherrschbar sein.
Liebe Grüsse, Mario Klessascheck
Hallo Herr Klessascheck,
inwieweit sind Handelsprodukte, bei welchen eine Standard-Gerätebatterie dem Produkt beigelegt wird, betroffen?
VG
Lieber Herr Kares,
Wenn Sie eine Batterie beifügen, dann sollten die Anforderungen an die Kennzeichnung, Austauschbarkeit und Entsorgung erfüllt sein. Als Händler müssten Sie die Konformitätsangaben der Batterie prüfen. Weitere besondere Anforderung konnte ich bisher nicht erkennen.
Liebe Grüsse, Mario Klessascheck
Sie schreiben zu den Übergangsfristen zur Entfernbarkeit von Batterien: „Die Anforderungen an die Entfernbarkeit und Austauschbarkeit gelten ab dem Tag des Inkrafttretens dieser Verordnung.“
In Art. 96, Satz 2 (a) kann man lesen, dass es für die Anforderungen des Art. 11 eine Übergangsfrist bis 18.02.2027 geben soll.
Können Sie das aufklären?
Vielen Dank vorab!
Gruß
Stefan Veitenhansl
Lieber Herr Veitenhansl,
vielen Dank für diesen wichtigen Hinweis. Der Fehler liegt bei uns. Ich habe die Übergangsfristen sogleich korrigiert. Nochmals vielen Dank für Ihr aufmerksames Lesen.
Liebe Grüsse, Mario Klessascheck
ich möchte das geschätzte Johner-Institut und die Leser auf folgendes Problem aufmerksam machen:
Der Begriff des „manufacturers“ ist im NLF eingeführt. Die Bezeichnungen „manufacturer“ und „Hersteller“ für diesen Akteur sind mannigfach etabliert. Nur leider ist bei der Übersetzung der Batterieverordnung eine Inkonsistenz eingebaut worden:
In der deutschen Version der Batterieverordnung (EUR-LEX) heisst der Wirtschftsakteur „manufacturer“ nicht „Hersteller“ sondern „Erzeuger“!
Überdies wurde das existierende Wort „Hersteller“ mit einer neuen Bedeutung belegt: Es entspricht nun dem englischen „Producer“ (was eine andere Rolle ist).
Ich kann allen betroffenen nur empfehlen, sich immer auf die konsistente englische Version zu beziehen und die deutsche Version zu meiden.
Ich halte das für einen bedauerlichen Fehler bei der Übersetzung. Man sollte nicht vergessen, dass die Übersetzungen rechtsgültig sind. Deutsche Gerichte beziehen sich üblicherweise auf deutsche Texte. Die Konsistenz mit ins Deutsche übersetzten Harmonisierten Normen wird ein weiteres Problem sein.
Lieber Herr Müller-Schöll,
vielen Dank für Ihren Beitrag. Ich darf Ihnen nachfolgend die Antwort meines Kollegen weiterleiten:
Lieber Herr Müller-Schöll,
Sie haben recht. Vielen Dank für den wichtigen Hinweis. Die Begriffsüberschneidung ist wirklich sehr verwirrend. Ob ein Übersetzungsfehler war, könnte man vermuten. Unsere Empfehlung wäre ebenfalls, mit der englischen Version zu arbeiten.
Liebe Grüsse
Mario Klessascheck
Herzliche Grüße
Tea Bodrusic
Werte Kollegen,
Zum Thema habe ich nun eine Antwort von der Europäischen Kommission erhalten, die ich unten anfüge. Die Übersetzung manufacturer = producer (abweichend von manufacturer = Hersteller) war offenbar „Absicht“. Es gibt wohl zwei verschiedene Übersetzungs-Communities bei der Kommission, die jeweils ihre eigene Linie pflegen. Dass Benutzer der Rechtsakte mit beiden in Kontakt kommen, war wohl nicht vorgesehen (!!??).
Hieraus kann man nur weiterhin nur die Empfehlung aufrechterhalten, konsequent beim englischen Text zu bleiben und die deutsche Übersetzung konsequent zu meiden.
************
Thank you for your message. This was already shared within our Batteries Regulation team, which fortunately contains a few German speakers.
However, unfortunately we have so far not identified a solution. The issue is that this was not a mistake, but a choice by the translators and the Member States involved in that translation. While you are correct that other EU (non-food) product legislation uses ‘Hersteller’ for ‘manufacturer’, there is also EU waste legislation that uses ‘Hersteller’ for ‘producer’, see in particular the Waste Framework Directive: https://eur-lex.europa.eu/eli/dir/2008/98/oj
Since waste legislation requires further rules at Member State level, the choice was made as it is now in the German (and some other language versions) of the Batteries Regulation.
Lieber Herr Müller-Schöll,
vielen Dank für Ihre wertvolle Ergänzung. Mit dem Begriff Erzeuger (Manufacturer) hat die EU das New Legislative Framework (NLF) erweitert. Der Hersteller, der aus dem NLF und der MDR bekannt war, wird nun Erzeuger genannt. Das bedeutet, dass MDR-Hersteller von Medizinprodukten in einem gewissem Ausmaß nun auch die erweiterten Herstellerpflichten im Sinne der Batterieverordnung übernehmen müssen.
Herzlichen Dank, Mario Klessascheck
Guten Tag Herr Klessascheck,
Sie haben zum Thema Austauschbarkeit der Batterien folgendes geschrieben:
„Für folgende Geräte kann das Austauschen der Batterie auf unabhängige Fachleute beschränkt bleiben:
Geräte, bei denen die Batterie nur funktionieren kann, wenn sie in die Gerätestruktur integriert ist (z. B. gemeinsames Gehäuse, ggf. Hörgeräte)“
Gibt es konkret aus der Verordnung das Beispiel zum Hörgeräte? Müssen diese durch Fachleute ausgetauscht werden? Wo steht bitte diese Behauptung in der Verordnung?
Vielen Dank
Liebe Ndengue,
Meine Aussage basiert auf Artikel 11, Absatz 2a. Dieser erlaubt eine Vereinfachung, wenn Produkte für den Einsatz in Umgebungen vorgesehen sind, in denen sie mit Flüssigkeiten in Kontakt kommen können – vorausgesetzt, sie sind so konstruiert, dass kein Kurzschluss an der Batterie entstehen kann. Würde ein Endanwender die Batterie selbst austauschen, wäre nicht mehr gewährleistet, dass das Produkt weiterhin die IP-X-Anforderungen erfüllt.
Die EU hat im Januar eine Leitlinie (C/2025/214) veröffentlicht. Hörgeräte werden darin als relevant für die Ausnahmeregelung in Artikel 11 Absatz 3 angesehen. Ich werde das Beispiel im Blogartikel entsprechend anpassen.
Liebe Grüsse, Mario Klessascheck