
Dieser Beitrag beschreibt, wann und weshalb Hersteller ein CE-Zeichen anbringen müssen, was man unter CE-Kennzeichnung versteht und was die Hersteller bis zu diesem Zeitpunkt unternehmen müssen.
Umgangssprachlich werden die Begriffe CE-Zeichen, CE-Kennzeichen und CE-Kennzeichnung synonym verwendet. Die MDR verwendet die Begriffe CE-Konformitätskennzeichnung und CE-Kennzeichnung.
Im Englischen heißt es CE marking bzw. CE marking of conformity.
1. Bedeutung des CE-Zeichens
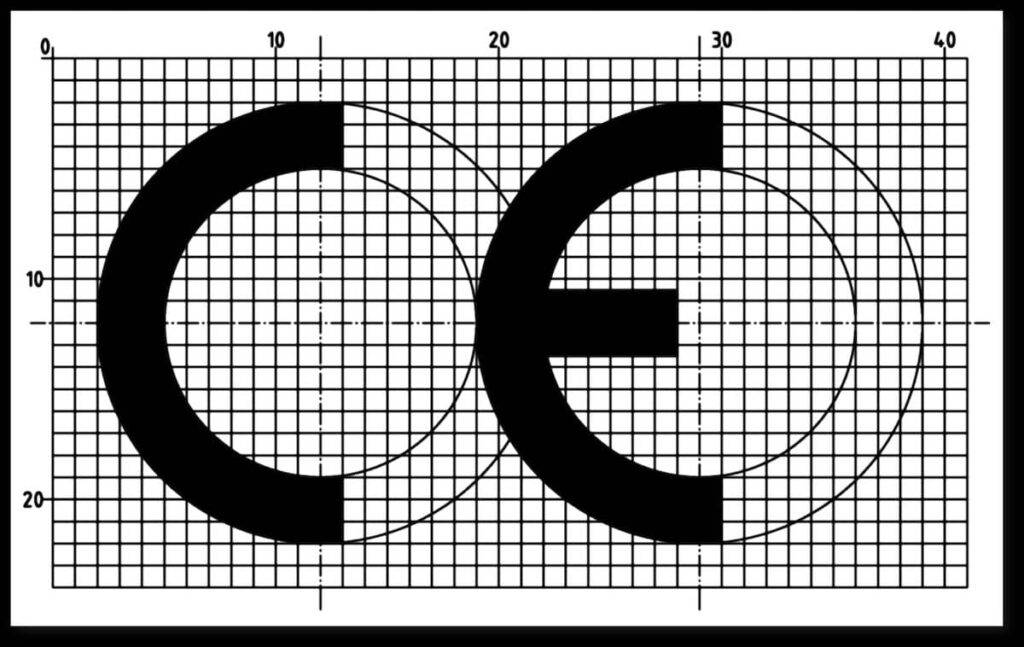
Abbildung 1 (links): In Anhang V von MDR und IVDR wird das Aussehen der CE-Kennzeichnung festgelegt.
Mit dem CE-Zeichen drücken Hersteller aus, dass ihr Produkt den Anforderungen einer europäischen Richtlinie oder einer EU-Verordnung entspricht.
Die Medizinproduktehersteller drücken mit der CE-Kennzeichnung die Konformität ihrer Produkte mit den EU-Verordnungen MDR bzw. IVDR aus.
Den Begriff CE-Audit gibt es nicht.
2. Anforderungen an die CE-Kennzeichnung
MDR und IVDR fordern von den Herstellern bezüglich der CE-Kennzeichnung das Folgende:
- Medizinprodukte müssen die CE-Kennzeichnung dauerhaft tragen. (Ausnahmen gibt es für Sonderanfertigungen und Prüfprodukte.)
- Das CE-Zeichen muss so wie in Abb. 1 dargestellt aussehen und mindestens 5 mm hoch sein.
- Falls beim Konformitätsbewertungsverfahren eine Benannte Stelle beteiligt wurde, muss deren Nummer neben dem CE-Zeichen stehen (s. Abb. 2).
- Die Medizinprodukte müssen die Anforderungen der Verordnungen (und ggf. weiterer Verordnungen und Richtlinien) erfüllen, bevor der Hersteller die Konformität erklären und die CE-Kennzeichnung anbringen darf.

3. Anbringen des CE-Zeichens bei Software
Auch Standalone-Software (Webseiten, Apps oder PC-Software) kann ein Medizinprodukt sein. Daher bedürfen diese Produkte ebenfalls einer CE-Kennzeichnung.
Lesen Sie mehr dazu, wann Software als Medizinprodukt zählt. In den allermeisten Fällen fällt diese Software dann in die Klasse IIa oder höher. Dann muss der Hersteller muss eine Benannte Stelle einbeziehen und deren Nummer auf dem CE-Zeichen angeben.
Es gibt nur noch wenig Klasse-I-Software.
Hersteller sollten das CE-Kennzeichen bei Standalone-Software, soweit sinnvoll und möglich, anbringen an bzw. bei:
- Start- bzw. Splash-Screen
- Hilfe, About
- Webseite selbst
- Begleitmaterialien, insbesondere Gebrauchsanweisung, Handbuch
Es besteht keine regulatorische Pflicht, das CE-Zeichen in einem Web- bzw. Appstore anzubringen.
Die Anforderungen von MDR und IVDR gelten unabhängig davon, ob das Angebot beispielsweise einer Webseite kostenlos ist oder nicht oder ob die Seite nur über ein Passwort zugänglich ist oder eben nicht. Damit gilt die Pflicht zur Anbringung der CE-Kennzeichnung ebenfalls unabhängig davon.
4. Der Weg zum CE-Zeichen
Die unten verlinkte Präsentation verschafft einen schnellen Überblick über die Schritte, die notwendig sind, um die CE-Kennzeichnung an Medizinprodukten anbringen und diese gesetzeskonform in den Markt bringen zu dürfen.
Klicken Sie auf den unteren Button, um den Inhalt von www.slideshare.net zu laden.
Beachten Sie auch den Artikel In 7 Schritten zum Medizinprodukt.
a) Übersicht über die Phasen
Der ganze Prozess bis zunm Erhalt des CE-Zeichens umfasst mehrere Phasen (s. Abb. 3).
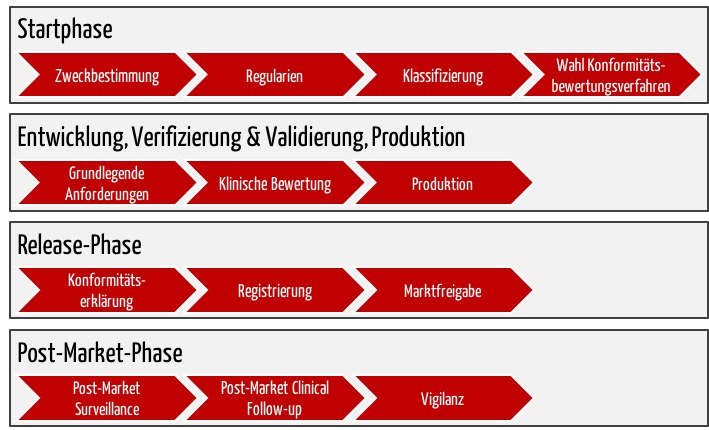
b) Startphase: Die Vorbereitungen zum CE-Zeichen
- Zweckbestimmung
Formulieren Sie die Zweckbestimmung Ihres Produkts. Legen Sie genau fest, ob und wie es der Diagnose, Therapie oder Überwachung von Krankheiten und Verletzungen dient, wer die vorgesehenen Benutzer sind und welches der vorhergesehene Benutzungskontext ist. - Regulatorische Anforderungen
Ermitteln Sie, welche EU-Verordnungen Ihr Medizinprodukt betreffen (Medizinprodukteverordnung MDR, IVD-Verordnung IVDR). - Klassifikation
Wenn es sich um ein Produkt handelt, das unter die MDR fällt, ermitteln Sie mithilfe der Klassifizierungsregeln im Anhang VIII die Klasse, in die Ihr Medizinprodukt fällt (I, IIa, IIb oder III).
Bei dieser Klassifizierung können wir Sie unterstützen (Kontakt aufnehmen). - Wahl des Konformitätsbewertungsverfahrens
Abhängig von der gewählten Klasse wählen Sie das Konformitätsbewertungsverfahren und müssen dann ggf. ein Qualitätsmanagementsystem etablieren, das Sie nach ISO 13485 zertifizieren lassen müssen.
Mit unserer Hilfe gelingt das den Firmen immer auf Anhieb.
Es gibt also keine „CE-Zertifizierung“ und kein „CE-Audit“, aber ggf. ein QM-Audit und ein ISO-13485-Zertifikat bzw. Anhangs-Zertifikat (gemäß den Anhängen der MDD oder MDR).
c) Entwicklungs-, Test- und Produktionsphase
- Grundlegende Anforderungen
Entwickeln Sie das Medizinprodukt gemäß den Vorgaben Ihres QM-Systems, falls dieses existiert. In jedem Fall muss es alle grundlegenden Anforderungen einhalten, z. B. den Anforderungen zum Risikomanagement, zu Software-Lebenszyklusprozessen, zur Gebrauchstauglichkeit und zur elektrischen Sicherheit.
Dies weisen Sie nach, indem Sie harmonisierte Normen einhalten und dies durch eine Technische Dokumentation belegen.
Diese Technische Dokumentation enthält folglich:- Risikomanagementakte (konform ISO 14971)
- Software-Akte (gemäß IEC 62304)
- Gebrauchstauglichkeitsakte (konform IEC 62366)
- Akte zur elektrischen Sicherheit (konform IEC 60601-1)
- Klinische Bewertung
Sie weisen im Rahmen der klinischen Bewertung mit klinischen Daten nach, dass Ihr Medizinprodukt keine ungewünschten Nebenwirkungen hat und den beabsichtigten Nutzen erfüllt. Falls es keine belastbaren Literaturdaten gibt, müssen Sie eine klinische Prüfung durchführen. - Produktion
Schließlich produzieren Sie, d. h. vervielfältigen Ihr Medizinprodukt.
d) Release-Phase inklusive CE-Kennzeichnung
- Konformitätserklärung
Erklären Sie die Konformität Ihres Produkts mit den grundlegenden Anforderungen. Dazu erstellen Sie eine Konformitätserklärung und bringen das CE-Zeichen auf Ihrem Produkt auf. - Registrierung
Registrieren Sie sich und Ihr Medizinprodukt in der EUDAMED bzw. beim BfARM. - Marktfreigabe
Erteilen Sie abschließend die Marktfreigabe und bringen Sie Ihr Produkt – mit CE-Zeichen – in Verkehr.
e) Post-Market-Phase
Die letzte Phase dauert so lange an, wie sich mindestens eines Ihrer Produkte im Markt befindet. In dieser Phase prüfen Sie kontinuierlich Rückmeldungen aus dem „Feld“, reagieren auf Zwischenfälle und aktualisieren Ihre Risikomanagementakte.
Lesen Sie hier mehr zum Thema Post-Market Surveillance und Vigilanz.

Von null auf konform in Rekordzeit!
Mit dem Auditgarant erstellen Sie in kürzester Zeit eine gesetzeskonforme technische Dokumentation für Ihr Medizinprodukt. Nach einem individuellen Onboarding mit unseren erfahrenen Berater:innen zeigen wir Ihnen genau, was Sie in welcher Reihenfolge erledigen müssen, um Ihr Produkt schnell in den Verkehr zu bringen.
5. Fazit und Zusammenfassung
Das CE-Zeichen anzubringen, zählt zu den leichteren Aufgaben, die Hersteller bewältigen müssen, um ihre Medizinprodukte gesetzeskonform in den Markt bringen und dort auch belassen zu dürfen. Der schwierige Teil besteht darin, die Voraussetzungen dafür zu schaffen, d.h. Konformität nachzuweisen. Dennoch muss auch diese CE-Kennzeichnung den gesetzlichen Anforderungen genügen.
Das Johner Institut unterstützt die Hersteller beim kompletten Prozess einschließlich dem Erstellen der technischen Dokumentation und der klinischen Bewertung.
Änderungshistorie
- 2023-08-22: Nur redaktionelle Änderungen (u.a. Rechtschreibfehler beseitigt)
- 2023-05-04: Artikel grundlegend überarbeitet, Anforderungen der MDR genauer beschrieben
- 2019-06-17: Erste Version des Artikels



Lieber Herr Johner,
ich möchte gerne etwas ergänzen, weil Sprachgebrauch und Wahrnehmung „da draußen“ so vielfältig sind, dass sonst der ein oder andere vielleicht verunsichert ist. Wohlgemerkt, Ihre Ausführungen oben sind natürlich richtig.
– Auch in der MDD (und den anderen Richtlinien) stehen explizite Anforderungen an das QM-System. Während eines Audits nach ISO 13485 werden diese Anforderungen direkt mit auditiert. Das wird durchaus manchmal als CE-Audit begriffen. Merke: So wichtige Dinge wie die SOP für das Vigilanz-System und der Sicherheitsbeauftragte werden in der Richtlinie bzw. dem MPG gefordert, nicht aber in der ISO 13485.
– Bei den Konformitätsbewertungsverfahren mit Beteiligung einer Benannten Stelle (BS) reicht man die Technische Dokumentation ein. Die BS prüft dann (off-site, wie man so schön sagt). Ich habe, auch wenn das sachlich falsch ist, auch in diesem Zusammenhang schon das Wort Audit gehört. Die Tätigkeit ist aber auch nicht gänzlich entfernt, die BS prüft die TD auf Konformität mit den Anforderungen der Richtlinie. Im Anschluss erhält man von der BS eine „Konformitätsbescheinigung“ oder „CE-Certificate“: Achtung, da habe ich schon je nach BS unterschiedlichstee Begriffe gesehen, nicht alle sind „richtig“, selbst das Verfahren und die Form und der Inhalt des Zertifikates unterscheiden sich.
Ohne dieses Zertifikat darf der Hersteller, abhängig vom Konf.-Bewert.-Verfahren die CE-Kennzeichnung dann doch nicht anbringen.
Also die BS prüft und stellt nachher ein Zertifikat aus, ohne das ich nicht weiterkomme. Der Volksmund mag das genauso benennen wie beim ISO-Zertifikat, selbst wenn es streng genommen nicht richtig ist.
– Seit der Änderung der Richtlinie in 2007 (2007/47), also effektiv seit 2010, haben die Benannten Stellen auch in jedem Audit eine Stichprobe aus den Technischen Dokumentation der Hersteller zu prüfen. Ein Audit, das die „CE-Dokumentation“ betrifft. Verkürzt ein „CE-Audit“?
Viele Grüße und eine gute Woche
Peter Knipp
Danke für die wertvollen Kommentare, lieber Herr Knipp!
Herzliche Grüße
Christian Johner
Lieber Herr Prof. Johner,
wir erstellen eine Softwarekomponente (62304 Klasse B), die beim Inverkehrbringer (das sind wir nicht) in ein Medizinprodukt integriert wird. Klar, dass der Inverkehrbringer das CE-Kennzeichen an sein Produkt anbringen muss. Ist es auch erforderlich, dass wir als Software-Lieferant ein Konformitätsbewertungsverfahren durchführen und das CE-Kennzeichen an unsere Software-Komponente anbringen müssen?
Beste Grüße
Karl Mösel
Sehr geehrter Herr Mösel,
wenn Sie nicht der Inverkehrbringer sind, müssen Sie gar nichts. Wenn Ihre Komponente selbst kein Medizinprodukt ist, sondern eben „nur“ eine Komponente“, dann dürfen Sie gar kein CE-Kennzeichen anbringen.
Hilft das? Falls nicht, haken Sie gerne nach.
Christian Johner
Lieber Herr Prof. Johner,
wie lange sind CE-Certificate unter der MDR bzw. IVDR gültig, d.h. in welchen Abständen gibt es Re-Zertifizierungen?
Besten Dank und viele Grüsse
Walter Bender
Sehr geehrter Herr Bender,
die Gültigkeit steht auf Ihren Anhangszertifikaten. Deren Gültigkeit ist aber auf maximal 2024 im Fall der MDR begrenzt.
Die Überwachungsaudits erfolgen jährlich, Re-Zertifizierungsaudits alle drei Jahre.
Beste Grüße, Christian Johner
Lieber Herr Prof. Johner,
momentan laufen auf dem deutschen Markt einige Testversionen von unserer SEMIC EyeScan App, die über eine Bildaufnahme des menschlichen Auges mit einem Smartphone innerhalb von 3 Minuten erkennen kann, ob ein Mensch mit COVID-19 infiziert ist. Meine Frage an Sie: brauchen wir hierzu in irgendeiner Form eine CE-Zulassung? Wir nutzen die Methode des Sterilen Rechnens https://www.semic.de/de/presse/d/semic-sidekick-nutzt-steriles-rechnen-zum-schutz-ihrer-daten und erfüllen damit die EU Datenschutzrichtlinien. Die Logik-Software wird auf dem Smartphone geladen und führt die Berechnungen durch. Es werden keine Daten aus dem Benutzergerät entnommen. Nachdem die Berechnung auf dem Endgerät abgeschlossen ist, löscht sich die Logik-Software automatisch.
Besten Dank für Ihre Antwort im Voraus!
W. Gruber
Sehr geehrter Herr Gruber,
wenn die Zweckbestimmung Ihrer App darin besteht, eine COVID-Infektion zu erkennen, dann ist das höchstwahrscheinlich ein IVD und Sie brauchen eine CE-Kennzeichnung. Eine Bereitstellung von Testversionen entspricht einer Inverkehrbringung (illegal) oder einer klinischen Prüfung bzw. klinischen Leistungsbewertung, was ebenfalls illegal wäre, wenn die entsprechenden sehr umfangreichen Anforderungen nicht erfüllt sind.
Fazit: Wenn ich Sie richtig verstehe, gibt es dringenden Handlungsbedarf.
Viele Grüße, Christian Johner
PS: Falls Sie Unterstützung benötigen, dann melden Sie sich.
Sehr geehrter Herr Johner,
Sie/ihr Institut schreiben/t in diesem Artikel, dass das CE Zeichen auf Begleitmaterialien angebracht werden muss. Handelt es sich nur um Begleitpapiere (instruction for use, Handbücher) oder auch beispielsweise Flyer und Broschüren, die das Produkt nicht begleiten? Die MDD und MDR geben leider keine klare Information in diesem Kontext.
Unser Auffassung zur Folge können CE Kennzeichnung auf Flyer u.U. verwirren, da diese als CE Kennzeichnung des Werbematerials als solches verstanden werden könnten.
Über eine zügige Antwort von ihnen würde ich mich sehr freuen, vielen Dank in diesem Zusammenhang auch an Sie und ihr Team für diese sehr guten, prägnant aufgearbeiteten Inhalte zum Thema MD. Für mich und mein Team ein ständiger Begleiter bei unser Arbeit rund um das Thema QM.
Mit freundlichen Grüßen
Kevin Küssner
Sehr geehrter Herr Küssner,
besten Dank für Ihren wichtigen Hinweis!
Sie haben Recht, dass ein CE-Zeichen auf dem Flyer keinen Sinn ergibt. Ich hatte daher geschrieben „Begleitmaterialien insbesondere Gebrauchsanweisung, Handbuch“. Aber Flyer fallen ggf. auch unter die Begleitmaterialien. Ich hatte zwar eher die „vom Hersteller gelieferten Informationen“ im Kopf. Aber das werde ich im nächsten Update klarstellen.
Sie haben mein „soll“ bereits als „muss“ interpretiert. Das ist mit der MDR inzwischen auch so, wie der Artikel 20 fordert. Das werde ich ebenalls schärfen.
Nochmals vielen Dank für Ihren Hinweis und Ihr wertschätzendes Feedback, über das ich mich freue!
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
können Produktzertifikate, die von einer Benannten Stelle für Medizinprodukte ausgestellt wurden von einem Unternehmen auf ein anderes übertragen werden? Wenn ein Unternehmen ein anderes Medizinprodukteunternehmen übernimmt, gelten dann die Zertifikate für das neue Unternehmen und dieses kann die Konformitätserklärung bezugnehmend auf diese Zertifikate ausstellen? Wie ist hier der Ablauf und was muss man beachten?
Danke und schöne Grüße,
Martina
Sehr geehrte Martina,
das ist eine spannende Frage. Danke dafür!
Um die Frage beantworten zu können, müsste ich das Konformitätsbewertungsverfahren kennen, für das das Zertifikat ausgestellt wurde. Auch die Frage, ob es Zertifikate unter der MDD oder MDR sind, müsste geklärt sein.
Nutzen Sie gerne das kostenlose Micro-Consulting, um eine passgenaue Antwort zu bekommen. Sie müssten uns dort nur kurze Hinweise zu diesen Fragen geben.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner, wir haben recherchiert und nirgends eine Antwort darauf gefunden: muss die Nummer der Benannten Stelle die gleiche Größe haben wie das CE-Zeichen? In Annex V steht „The various components of the CE marking shall have substantially the same vertical dimension […]“. Zählt die Nummer als Komponente oder nicht?
Viele Grüße
Kim Heimhuber
Liebe Frau Heimhuber,
meine Kollegin Astrid Schulze hat darauf die folgende Antwort gegeben:
die Nummer der Benannten Stelle muss nicht die gleiche Größe des
CE-Kennzeichens haben. Die von Ihnen zitierte Textpassage aus dem Anhang V
bezieht sich nur auf das CE-Kennzeichen selbst, also die Buchstaben C und E.
Mit besten Grüßen
Astrid Schulze
Guten Tag,
vielen Dank für die vielen hilfreichen Artikel.
Ich erlaube mir, eine kurze Frage zu stellen.
Gibt es die Möglichkeit, das CE-Zeichen nicht direkt auf dem Produkt anzubringen (Platzmangel oder ähnliches).
Bei uns wäre dies vor allem für die Zubehörteile (Nasenadapter für Pumpdosieraufsatz) relevant.
Vielen Dank und herzliche Grüße
Beate Jenne
Liebe Frau Jenne,
ja diese Möglichkeit gibt es. Bei manchen Produkten ist es nicht sinnvoll und würde die Sicherheit beeinträchtigen. Auch Platzmangel wird als Grund akzeptiert. Dann muss das CE Kennzeichen auf der Verpackung und in der Gebrauchsanweisung zu finden sein.
Regulatorische Grundlagen: Art. 2,13 MDR: 13. „Kennzeichnung“ bezeichnet geschriebene, gedruckte oder grafisch dargestellte Informationen, die entweder auf dem Produkt selbst oder auf der Verpackung jeder Einheit oder auf der Verpackung mehrerer Produkte angebracht sind;
Artikel 20, (3), MDR: (3) Die CE-Kennzeichnung wird gut sichtbar, leserlich und dauerhaft auf dem Produkt oder auf seiner sterilen Verpackung angebracht. Ist diese Anbringung wegen der Beschaffenheit des Produkts nicht möglich oder nicht sinnvoll, wird die CE-Kennzeichnung auf der Verpackung angebracht. Die CE-Kennzeichnung erscheint auch in jeder Gebrauchsanweisung und auf jeder Handelsverpackung.
Mit besten Grüßen
Astrid Schulze
Sehr geehrtes Johner Institut!
Ich habe eine Frage zur Abbildung 2 – dem CE-Zeichen inkl. der Nummer der Benannten Stelle. Kann diese Nummer theoretisch auch unterhalb des CE-Zeichens geschrieben werden (z.B. aus Platzmangel)? Ich habe hierzu keine genauen Angaben im Artikel 20 oder im Anhang 5 finden können.
Mit freundlichen Grüßen,
Kathrin
Sehr geehrte Kathrin,
die Nummer muss gemäß 93/465/EWG Anhang I, B 2. g) hinter dem CE-Kennzeichen stehen. Eine Ausnahme ist mir nicht bekannt.
Beste Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
muss die Nummer der Benannten Stelle auch nicht die gleiche Größe wie das CE-Zeichen haben (siehe Directive 2014/53/EU article 20.3)?
In diesem Falle müsste Ihr Beispiel im Punkt 2 berichtigt werden.
Beste Grüße
Sehr geehrter Herr Cecek,
herzlichen Dank für Ihren wichtigen Hinweis!
Wie Sie schreiben enthält die Richtlinie 2014/53/EU zu den Funkanlagen(!) einen Hinweis auf die Schriftgröße. Die MDR und IVDR enthalten diese Anforderung nicht und verweisen auf die Verordnung 765/2008 („Akkreditierung und Marktüberwachung im Zusammenhang mit der Vermarktung von Produkten“). Letztere Verordnung wiederum gibt (wie die MDR/IVDR) nur die Mindestgröße des CE-Zeichens vor, nicht die Größe der Zahlen, welche die Nummer der Benannten Stelle repräsentiert. Vielleicht ist das der Grund, weshalb die CE-Kennzeichnung vieler Medizinprodukte eine kleinere Nummer enthalten.
Eine Pflicht kann ich somit nicht erkennen. Aber mit einer Angleichung der Größe von Zahlen und CE-Zeichen ist man auf der sicheren Seite.
Nochmals besten Dank!
Viele Grüße, Christian Johner
Guten Tag,
wir würden gerne ein Medizinprodukt (Klasse Is) in der Ukraine vermarkten. Wir haben es in der EU zugelassen. Wissen Sie, ob es zusätzlich notwendig ist, es in der Ukraine zu registrieren und wenn ja, wie? Unser Produkt wird als Kombinationsprodukt mit einem Arzneimittel, welches in der Ukraine produziert wird, vermarktet.
Mit freundlichen Grüßen
Dr. Ann-Kathrin Desch
Guten Tag Frau Desch,
ein CE-gekennzeichnetes Medizinprodukt kann nicht ohne Weiteres in die Ukraine verkauft werden. Es muss den lokalen Regulierungsvorschriften entsprechen und separat zugelassen werden. Wenn das Produkt der MDR entspricht, gibt es einen verkürzten Weg. Zusätzlich benötigen Sie einen Repräsentanten vor Ort. Bitte beachten Sie, dass es möglicherweise zusätzliche Anforderungen für Kombinationsprodukte gibt. Melden Sie sich gerne bei uns, wenn Sie Unterstützung bei der Recherche der lokalen Anforderungen und der Zulassung benötigen.
Herzliche Grüße,
Manuela Reinhold
Sehr geehrtes Johner Institut!
Ich habe eine Frage bezüglich des Anbringens des CE-Zeichens auf Werbematerialien. Ist es zulässig, ein CE-Zeichen auf Werbematerialien abzubilden, um darzustellen, dass die Produkte CE-zertifiziert sind? Wenn ja, müsste dann das CE-Symbol mit der zugehörigen Notified Body Nummer abgebildet werden?
Mit freundlichen Grüßen,
Kathrin
Liebe Kathrin,
gemäß Artikel 20 der MDR sind Sie sogar verpflichtet, das CE-Kennzeichen inklusive der Kennnummer der zuständigen Benannten Stelle in den Werbematerialien abzubilden.
Liebe Grüße,
Manuela Reinhold
Sehr geehrtes Johner-Team!
Ich habe einen Kunden in den USA, der mich gebeten hat, für die Produkte (die in Europa als Klasse IIa CE-zertifiziert sind) die er kauft, auf dem Label das CE zu entfernen. Grundsätzlich dürfte das kein Problem darstellen, weil diese ja in die USA exportiert werden und er diese nur in den USA vertreibt. Ist meine Annahme richtig?
Mit freundlichen Grüßen,
Michael St.
Lieber Herr Steiner,
ich kann Ihrer Annahme folgen. Die CE-Kennzeichnung ist für die Inverkehrbringung von Medizinprodukten innerhalb der Europäischen Union gefordert. Diese Kennzeichnung berechtigt nicht dazu, die Produkte in die USA zu vertreiben. Dafür müssen die Produkte den dortigen lokalen Anforderungen an Medizinprodukte, deren Zulassung und Kennzeichnung entsprechen. Sie können also das CE-Kennzeichen von den Produkten entfernen, insofern diese ausschließlich für den Vertrieb in den USA bestimmt sind.
Herzliche Grüße,
Manuela Reinhold