
Medizinprodukte basieren zunehmend auf Closed-Loop-Systemen. Diese „geschlossenen Regelsysteme“ finden bereits in der Medizinprodukteverordnung MDR Erwähnung. Ein Beispiel ist ein System aus einer Insulinpumpe, die von einem Gerät mit Glukosesensor gesteuert wird.
Sie erfahren in diesem Artikel, was Closed-Loop-Systeme sind, wo sie in der Medizin zum Einsatz kommen und welche regulatorischen Anforderungen sie erfüllen müssen. Der Beitrag verrät Ihnen auch, wie Sie beim Kauf der Norm IEC 60601-1-10 einige Hundert Euro sparen können.
1. Ziele von geschlossenen Regelsystemen (Closed-Loop-Systeme)
Closed-Loop-Systeme haben das Ziel, eine Regelgröße auf einem definierten Wert (Führungsgröße, Sollwert) zu halten, obwohl eine Störgröße das System beeinflusst.
Beispielsweise soll die Raumtemperatur eines Zimmers (Regelgröße) auf einem bestimmten Wert (Führungsgröße) gehalten werden, obwohl die Umgebungstemperatur das Zimmer abwechselnd aufheizt oder abkühlt (Störgröße).
Das besondere Merkmal eines geschlossenen Regelkreises bei ME-Geräten (medizinisch-elektrischen Geräten) ist die Messung einer physiologischen Variablen (Regelgröße), die dazu dient, die Zufuhr von Energie oder Stoffen (über ein Stellglied) anzupassen, um die physiologische Variable auf ein Ziel zu steuern oder zu halten.
Die kanadischen Behörden definieren Closed-Loop-Systeme wie folgt:
“Closed-Loop-System in respect of a medical device, means a system that enables the device to sense, interpret and treat a medical condition without human intervention.”
Diese Definition ist jedoch nicht präzise:
- Es muss klar sein, dass das Gerät eine Krankheit nicht nur erkennen und behandeln kann. Vielmehr muss diese Behandlung auf Basis der Erkennung erfolgen.
- Bei einem „echten“ Closed-Loop-System würde der Zyklus Messen – Handeln – Messen mehrfach durchlaufen. Das scheint bei dieser Definition nicht zwingend vorausgesetzt zu sein.
Eine bessere Definition liefert die FDA (weiter unten in Kapitel 4.b). Zunächst jedoch zurück zur Funktionsweise eines Regelkreises.
2. Funktionsweise von Closed-Loop-Systemen
a) Einführung
Geschlossene Regelsysteme nutzen die Regelgröße (Ist-Wert; in obigem Beispiel die tatsächliche Zimmertemperatur), um deren Differenz von der Führungsgröße zu bestimmen. Diese Differenz versucht ein Regler zu minimieren. Dazu hat er die Möglichkeit, eine Stelleinrichtung (auch Stellglied genannt) über eine Steuergröße zu beeinflussen. Im Fall unserer Heizung sendet der Regler eine Nachricht an das Ventil. Dadurch verändert sich die Ventilöffnung (Steuergröße) und damit die Menge des heißen Wassers im Zulauf des Heizkörpers.
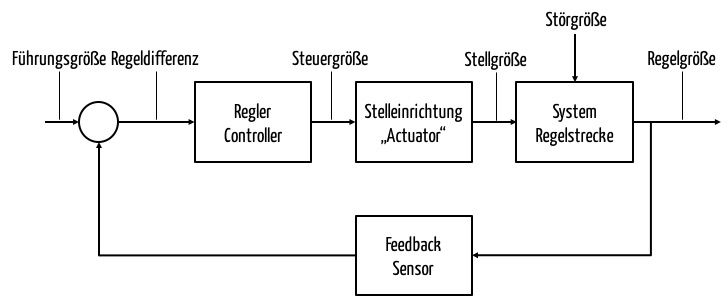
b) Typen an Reglern
Abhängig von den Anforderungen an die Güte der Regelung und abhängig von den technischen und finanziellen Möglichkeiten wählt man unterschiedliche Typen von Reglern.
Zwei- und Mehrpunkt-Regler
Zweipunkt-Regler zählen zu den primitivsten Typen von Reglern. Bei ihnen kennt die Stellgröße nur zwei Einstellungen, z. B. „ein“ und „aus“. Zweipunkt-Regler werden z. B. eingesetzt, um den Füllstand von Gefäßen zu regeln. Wird ein gewisser Füllstand (die Regelgröße) unter- oder überschritten, schaltet eine Pumpe ein oder aus.
Bei Mehrpunktreglern gibt es entsprechend nicht nur zwei „Schaltstufen“, sondern mehrere.
P-Regler
Bei einem P-Regler ändert sich die Stellgröße proportional zur Regeldifferenz. Bei einer Heizung wäre – innerhalb gewisser Grenzen – die Ventilöffnung umso größer, je niedriger die Raumtemperatur im Vergleich zur Solltemperatur ist – und zwar proportional größer.
I-Regler
Die I-Regler betrachten nicht (nur) die Regeldifferenz, sondern deren Integral. Das heißt: Je länger die Regeldifferenz vorliegt, umso stärker greifen die Regler ein und ändern die Stellgröße entsprechend. Damit sind I-Regler die langsameren, dafür aber genaueren Regler.
Weitere Regler
Regelungstechniker unterscheiden viele weitere Typen, z. B. den D-Regler, sowie Kombinationen dieser Regler, z. B. die PI-, PD- und PID-Regler. Eine Übersicht über Regler erhalten Sie hier.
Beispiele für geschlossene Regelsysteme
a) Technologie, Ökologie, Ökonomie
In der Technik, Ökologie und Ökonomie gibt es zahllose Beispiele für geschlossene Regelsysteme:
- Tempomat bei Autos
- Automatische Bewässerung von Blumenkästen
- Höhe des Wildbestands geregelt durch Abschuss und Hege
- Zinsanpassung zur Steuerung der Inflation
Besonders im letzten Fall werden die Herausforderungen deutlich, die Regler bei großen und unvorhersehbaren Störgrößen bewältigen müssen. Dies gilt insbesondere, wenn die Störgrößen selbst durch die Regelgröße beeinflusst werden.
b) Medizin
Der menschliche Körper ist ein hochkomplexes Steuerungssystem, das viele physiologische Parameter regelt. Dazu zählen u. a. die Körpertemperatur, die Herzfrequenz, die Atmung, der Blutzuckerspiegel, der pH-Wert oder die Pupillengröße der Augen.
Im Gegensatz zum einfachen Beispiel der Heizung muss der Körper viele Regelgrößen gleichzeitig beherrschen, die zudem abhängig voneinander sind und ständig geänderten Führungsgrößen genügen sollen. Beispielsweise hebt der Körper bei Krankheit die Führungsgröße Temperatur an (Fieber) oder erhöht bei Anstrengung den Sollwert für Atem- und Herzfrequenz.
c) Medizintechnik
Die Medizintechnik nutzt geschlossene Regelkreise in vielfacher Weise.
- Automatisierte Narkosesysteme
- Systeme zur Stabilisierung der Hämodynamik (des Blutdrucks)
- Neurostimulatoren
- Infusionspumpe für Muskelrelaxantien
- Herzschrittmacher
Dabei lassen sich drei wesentliche Typen unterscheiden:
- Interne Regelkreise
Interne Regelkreise zeichnen sich dadurch aus, dass es keine nach außen sichtbare Regelgröße gibt. Beispielsweise steuert ein Regelkreis einen Lüfter, um die Temperatur innerhalb des Geräts nicht über einen Maximalwert steigen zu lassen. - Externe „technologische“ Regelkreise
Bei diesem Typ steuert der Regelkreis ein äußeres Verhalten des Medizinprodukts, typischerweise an einem Anwendungsteil. Zu den Beispielen zählen die Drehzahlsteuerung einer Knochenbohrmaschine oder der Strom bei einem HF-Chirurgiegerät, den das Gerät abhängig vom elektrischen Widerstand des Patienten steuert. - Externe „physiologische“ Regelkreise
Diese Form von Regelkreisen steuert physiologische Parameter. Beispiele hierzu sind: Festlegung der Dosis eines Narkosemittels abhängig von der Narkosetiefe, der Ultrafiltrationsrate eines Dialysegeräts abhängig von der Menge des Bluts oder der Insulindosis einer (implantierbaren) Insulinpumpe abhängig vom Blutzuckergehalt des Patienten (Beispiel).
Viele autonome Systeme nutzen einen oder mehrere geschlossene Regelkreise und zählen daher als Closed-Loop-Systeme.
4. Regulatorische Anforderungen an Closed-Loop-Systeme
a) Anforderungen der MDR
Grundlegende Anforderungen
Die MDR geht bei den grundlegenden Sicherheits- und Leistungsanforderungen nicht speziell auf Closed-Loop-Systeme ein. Vielmehr gelten die Anforderungen, die Sicherheit der Patienten nicht zu gefährden und nur vertretbare Risiken (gemessen am Nutzen) zu akzeptieren, für alle Medizinprodukte.
Zwar nicht spezifisch, aber zumindest relevant für geschlossene Regelsysteme ist die folgende Anforderung der MDR:
Diagnostische Produkte und Produkte mit Messfunktion werden so ausgelegt und hergestellt, dass auf der Grundlage geeigneter wissenschaftlicher und technischer Verfahren ausreichende Genauigkeit, Präzision und Stabilität für die Zweckbestimmung des Produkts gewährleistet sind. Der Hersteller gibt die Genauigkeitsgrenzen an.
MDR Anhang I 15.1
Alle Closed-Loop-Systeme basieren auf Messungen; viele haben auch Messfunktionen im Sinn der MDR.
Ebenfalls relevant sind die Vorgaben an Produkte, zu deren Bestandteilen programmierbare Elektroniksysteme gehören, und Software:
Produkte, zu deren Bestandteilen programmierbare Elektroniksysteme, einschließlich Software, gehören, oder Produkte in Form einer Software werden so ausgelegt, dass Wiederholbarkeit, Zuverlässigkeit und Leistung entsprechend ihrer bestimmungsgemäßen Verwendung gewährleistet sind. Für den Fall des Erstauftretens eines Defekts sind geeignete Vorkehrungen zu treffen, um sich daraus ergebende Risiken oder Leistungsbeeinträchtigungen auszuschließen oder sie so weit wie möglich zu verringern.
MDR Anhang I 17.1
Der Beweis der Wiederholbarkeit und Zuverlässigkeit stellt manche Hersteller von Closed-Loop-Systemen vor eine Herausforderung. Darauf kommt dieser Artikel weiter unten zu sprechen.
Klassifizierung: MDR Regel 22
Die MDR geht nur ein einziges Mal auf Closed-Loop-Systeme ein, nämlich mit den Klassifizierungsregeln in Anhang VIII. Regel 22 besagt:
Aktive therapeutische Produkte mit integrierter oder eingebauter diagnostischer Funktion, die das Patientenmanagement durch das Produkt erheblich bestimmt, wie etwa geschlossene Regelsysteme oder automatische externe Defibrillatoren, gehören zur Klasse III.
MDR Anhang VIII Regel 22
Dieser Satz bedarf einiger Kommentare:
- Unangemessene Klassifizierung
Diese Regel macht Medizingeräte wie Dialysemaschinen und Beatmungsgeräte zu Klasse-III-Produkten. Ob das wirklich die Absicht der Kommission war, darf bezweifelt werden. Diese Klassifizierung wäre abwegig. - Missverständnis dessen, was ein Closed-Loop-System ist?
Automatische externe Defibrillatoren (AID) sind nicht notwendigerweise Closed-Loop-Systeme – auch wenn sie „automatisch“ sind. Die AID bestimmen zwar automatisch das EKG-Signal des Patienten und entscheiden daraufhin, ob und welche Form der Defibrillation verwendet wird. Aber ein Feedback-Loop findet nicht statt. Es sei denn, das EKG-Signal war die Folge eines vorausgegangenen – offensichtlich nicht erfolgreichen – Versuchs.
Man muss aus dem Text nicht schlussfolgern, dass der Gesetzgeber AID zur Klasse der geschlossenen Regelsysteme zählt. Aber ob er wirklich verstanden hat, was ein Closed-Loop-System ist, kann diskutiert werden. Die Wahl eines Herzschrittmachers als Beispiel wäre nachvollziehbarer gewesen (Artikel dazu). - Es sind nicht alle geschlossenen Regelsysteme gemeint
Die Kommission wünscht wahrscheinlich nicht, alle geschlossenen Regelsysteme in die Klasse III einzuteilen. Vielmehr – so die Vermutung – möchte sie nur die geschlossenen Regelsysteme einbeziehen, die wir weiter oben als „externe physiologische Regelkreise“ bezeichnet haben. Das lässt sich daraus schließen, dass eine diagnostische Funktion eingebaut – und wahrscheinlich Teil des Regelkreises – sein muss. - Integriert versus eingebaut
Worin der Unterschied zwischen einer integrierten und einer eingebauten Funktion liegt, erschließt sich auch beim Lesen des englischen Textes („incorporated or integrated“) nicht unmittelbar.
Lesen Sie hier mehr zum Thema Klassifizierung von Software durch die MDR.
b) Anforderungen der FDA an Closed-Loop-Systeme
Fast zwei Jahre nach dem Entwurf einer Leitlinie speziell für Medizinprodukte zu geschlossenen Regelkreisen hat die FDA die finale Version dieses Guidance-Dokuments veröffentlicht.
Anwendungsbereich
Diese Leitlinie betrifft allerdings nur „physiological closed-loop controlled“ Medizinprodukte (PCLC-Produkte). Das sind Medizinprodukte, die folgende Bedingungen erfüllen:
- Die Produkte nutzen Sensoren, die physiologische Parameter messen.
- Sie verwenden Aktoren und Algorithmen, die, basierend auf den physiologischen Messwerten, physiologische Parameter steuern.
- Die Steuerung erfolgt über die Zuführung oder den Entzug von Energie oder Materialien (z. B. Medikamente, Flüssigkeiten, Gase).
Hersteller sollten diese Leitlinie in zwei Fällen beachten:
- Zulassungen von Medizinprodukten nach PMN/510(k), De Novo, PMA und HDE
- Damit verbundene Q-Submission Meetings mit der FDA
Die FDA weist darauf hin, dass insbesondere für aktive Implantate (wie Neurostimulatoren) weitere Anforderungen zu berücksichtigen sind. Die Leitlinie soll keine Empfehlungen für die Anwendung des Machine Learning geben.
Definition
Die FDA definiert die PCLC-Produkte:
“a medical device that automatically adjusts or maintains a physiologic variable(s) (i.e., the controlled physiologic variable) through delivery or removal of energy or article (e.g., drugs, or liquid or gas regulated as a medical device) using feedback from a physiologic-measuring sensor(s)”
Beispiele
Weiterhin nennt die FDA konkrete Beispiele für Produkte bzw. Funktionen, die als PCLC-Technologie betrachtet werden können:
- Narkosegasgeräte, die automatisch den Anteil des eingeatmeten Narkosemittels als Reaktion auf eine Endtidalgasmessung steuern
- Mechanische Beatmungsgeräte, die die Einstellungen (z. B. Anteil des eingeatmeten Sauerstoffs) als Reaktion auf eine Messung der Sauerstoffsättigung des Patienten automatisch anpassen
- Automatische Insulinpumpen, die die Insulinabgabe auf der Grundlage eines Blutzuckerwertes steuern
- Neu: Hypo- und Hyperthermie-Systeme, die die Temperatur automatisch in Abhängigkeit der am Körper des Patienten gemessenen Temperatur anpassen
Die FDA empfiehlt zudem, dass Hersteller die Pre-Submission Meetings oder Anfragen nutzen sollen, wenn sie unsicher sind, ob ihre klinische Funktion ein PCLC-System ist. Die Behörde liefert z. B. detailliertere Rückmeldungen zum klinischen Studiendesign.
Die FDA referenziert ebenfalls die IEC 60601-1-10 Ed. 1.2:2020, die im Anhang A weitere Beispiele für PCLC-Systeme nennt.
Wichtigste Anforderungen
Die FDA empfiehlt den Herstellern, die technischen Komponenten und Spezifikationen ihres PCLC-Geräts so zu beschreiben, wie in der IEC 60601-1-10:2020 angegeben (z. B. Abschnitt 8.2.2 „Gerätespezifikationen“).
Die FDA formuliert die Anforderungen an PCLC-Geräte in den Kapiteln V bis VII.
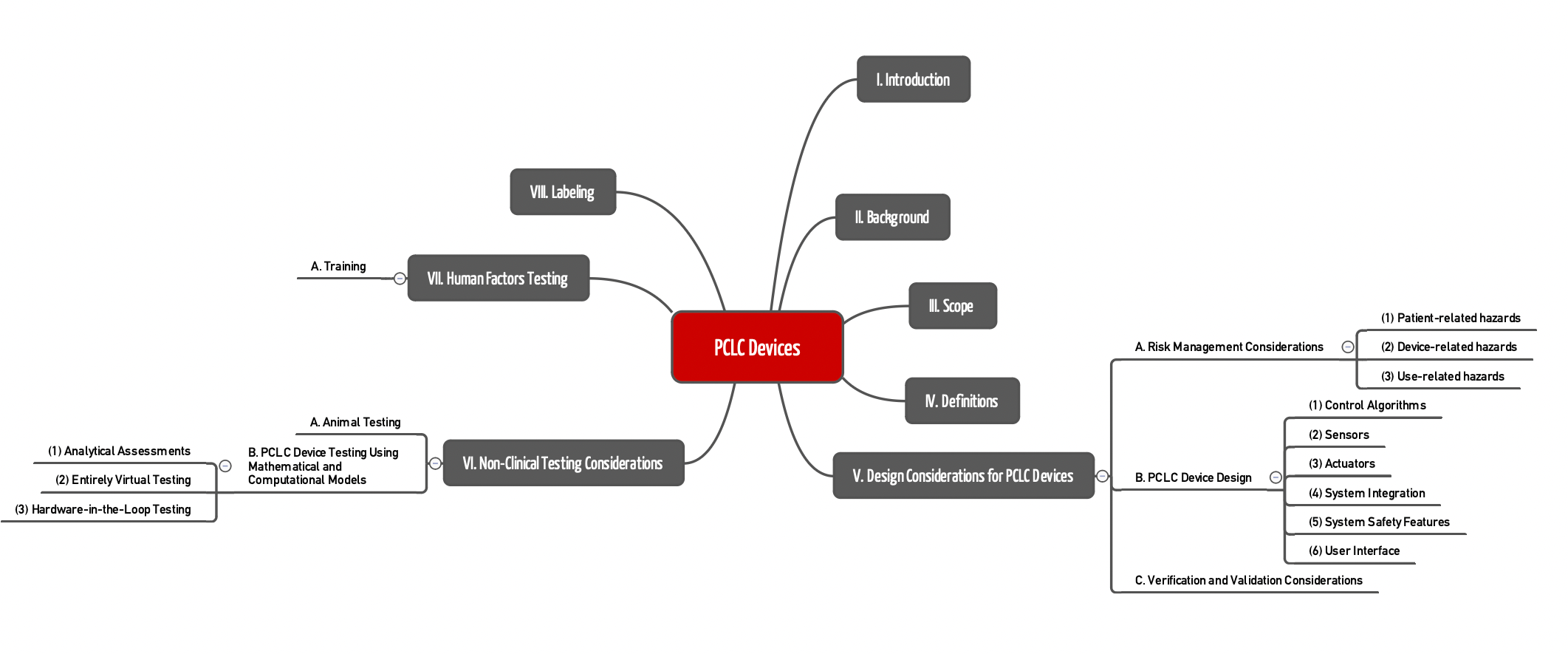
Kapitel V.A
Beim Risikomanagement (Kapitel V. A.) nennt die Leitlinie einige Gefährdungen und Risiken, die die Hersteller berücksichtigen sollten:
- Patientenbezogene Gefährdungen
Die Reaktion eines Patienten auf die Energie oder Substanz, die zugeführt oder entzogen wird, ist ein entscheidender Faktor, der bei der Auslegung zu berücksichtigen ist. Diese Reaktion kann von Patient zu Patient und auch bei einem einzelnen Patienten unterschiedlich sein. Es müssen etwa Störungen durch andere Therapien berücksichtigt werden. Dementsprechend sollen in der Risikoanalyse folgende Aspekte betrachtet werden:- Vorgesehene Patientengruppe (Variabilität der physiologischen Reaktion zwischen Patienten)
- Kontraindikationen für den Einsatz der PCLC-Technologie
- Umgebung, in der das Gerät eingesetzt werden soll
- Identifizierung von Unterschieden zwischen der derzeitigen Behandlung nach anerkannten Praxisleitlinien und der Methode, die von dem PCLC-Gerät angewendet werden soll
- Nutzungsbezogene Gefährdungen
Neben den üblichen Nutzungsfehlern betont die FDA Gefährdungen, die dadurch entstehen können, dass die Anwender dem System zu sehr vertrauen, eigene Fähigkeiten verlieren oder mit der Technologie überfordert sind. - Gerätebedingte Gefährdungen
Die Hersteller sollten die Unsicherheiten in ihrem Systemdesign sowie die vorhersehbaren funktionellen Störungen in der klinischen Umgebung und im Arbeitsablauf identifizieren und klar beschreiben, zum Beispiel:- Signalrauschen
- Unzureichende Sensorgenauigkeit bzw. Drift oder Ausfall
- Latenz- und Verzögerungszeiten
- Kommunikationsfehler zwischen Gerätekomponenten und Fehler innerhalb einer integrierten klinischen Umgebung
- Cybersecurity-Bedrohungen und -Schwachstellen
Die Leitlinie bezieht neu auch Elemente einer Integrierten Klinischen Umgebung ein, wie sie in der ASTM-Norm F-2761: „Medical Devices and Medical Systems — Essential safety requirements for equipment comprising the patient-centric integrated clinical environment (ICE) — Part 1: General requirements and conceptual model“ beschrieben sind.
Das ist insbesondere dann wichtig, wenn Patienten durch die Schaffung eines umfassenden medizinischen Systems, in das auch andere verteilte Systeme integriert sind, behandelt werden.
Kapitel V.B
Im Kapitel V. B. zum Design der Produkte gibt die FDA Empfehlungen zum Entwurf der verschiedenen Komponenten. Dabei verweist sie regelmäßig auf die Vorgaben der IEC 60601-1-10. Die Anforderungen sind teilweise relativ „high-level“, wie etwa die folgende:
„Control algorithms for all modes should be designed to meet their performance 539 specifications during reasonably foreseeable disturbances.”
Hilfreicher sind die Hinweise der FDA zum Design bzw. zur Auswahl der Komponenten. Beispiele sind
- die Linearität und die Latenz von Sensoren,
- die Genauigkeit und die physikalischen Grenzen von Aktoren sowie
- die physische Beschränkung bei den Reglern (z. B. der zuführbaren Energie) und Rückfallmöglichkeiten bei Fehlern.
Auch das „Data Logging“ und der Einsatz von Alarmen zählen zu den Empfehlungen der Behörde.
Kapitel V.C.
Das Kapitel V. C. beinhaltet eine umfassende Liste der Aspekte, die validiert bzw. verifiziert werden sollen. Das Kapitel beschreibt auch, wann eine Verifikation oder eine Validierung notwendig ist. Die Verifikation betrifft beispielsweise sämtliche Tests im Hinblick auf die Spezifikation. Ein Beispiel ist die Prüfung eines Sensors, der die Anforderungen des PCLC-Systems erfüllen muss. Die Validierung fokussiert auf Aspekte wie die Gestaltung der Benutzerschnittstelle. Die in diesem Abschnitt aufgeführte Liste dient somit gleichzeitig als eine Checkliste für den Umfang der zu erstellenden Spezifikationen.
Kapitel VI
In diesem Abschnitt geht das Dokument erneut auf die präklinischen Tests ein, die ein integraler Bestandteil der Verifikations- und Validierungsaktivität (V&V) sind. Kapitel VI unterstreicht die Notwendigkeit zusätzlicher Tests für PCLC-Systeme aufgrund ihrer besonderen Merkmale, welche in der Medizinprodukteprüfung berücksichtigt werden müssen.
Den Herstellern wird empfohlen, eine systematische Methode zur Entwicklung von Störungs- und Unsicherheitsszenarien für Stresstests des PCLC-Systems zu in Betracht zu ziehen, um sicherzustellen, dass diese unter klinisch relevanten Worst-Case-Bedingungen getestet werden.
- Tierversuche
Hersteller, die Tierversuche planen, um unter Worst-Case-Bedingungen zu testen, sollten bei der Planung der Studie die aufgeführten Aspekte in dem Abschnitt berücksichtigen. - Verwendung mathematischer und rechnerischer Modelle
Da Testen in allen klinisch relevanten Szenarien anhand von Tierversuchen und/oder klinischen Studien möglicherweise nicht machbar ist, empfiehlt die FDA in Ergänzung die Verwendung mathematischer und numerischer Modelle. Die Bewertung eines PCLC-Systems anhand eines Patientenmodells kann eine Alternative bzw. Ergänzung zu Tierversuchen und/oder klinischen Studien sein. Computermodelle, die für den Entwurf oder die Validierung eingesetzt werden, müssen auf ihre Vorhersagefähigkeit überprüft werden. Dabei verweist die FDA auf entsprechende Guidance-Dokumente und die im ASME V&V 40 beschriebenen Best Practices.
Lesen Sie hier mehr zu Computerbased Modeling and Simulation, dem zugehörigen FDA-Dokument sowie dem ASME V&V 40.
Kapitel VII
Wenig Neues enthält das siebte Kapitel. Dass die Leitlinie der FDA beim Human Factors and Usability Engineering zu beachten ist, liegt auf der Hand. Viele spezifische Gefährdungen wie den „Automation Bias“ nennt das Dokument bereits in vorangegangenen Abschnitten.
Kapitel VIII
Die FDA empfiehlt, die Dokumentation gemäß den Kapitel 5.1 „Gebrauchsanweisung“ und Kapitel 5.2 „Technische Beschreibung“ in der IEC 60601-1-10 aufzubauen und stellt klar, welche weiteren Aspekte die Hersteller beschreiben müssen, beispielsweise:
- Komponenten wie Sensoren und Aktoren mit jeweiligen Leistungsspezifikationen
- Alarme
- Funktionsweise
- Gesteuerte physiologische Parameter
- Voraussetzungen für den Einsatz
- Vorhandene bzw. notwendige Fallbacks
- Bedienung des Produkts
c) IEC 60601-1-10
Übersicht
Die IC 60601-1-10 trägt den deutschen Titel „Anforderungen an die Entwicklung von physiologischen geschlossenen Regelkreisen“. Sie wendet sich an medizinisch elektrische Geräte und zählt als Ergänzungsnorm zur Basisnorm, der IEC 60601-1.
Die IEC 60601-1-10 definiert (ähnlich wie die FDA) ein PCLC eher technisch über die Parameter eines Regelkreises. Im Prinzip sind beide Definitionen ähnlich, wenn auch nicht wortgleich.
Genau wie beim Guidance-Dokument der FDA (siehe weiter unten) beschränkt sich der Anwendungsbereich auf Medizinprodukte, die physiologische Parameter steuern.
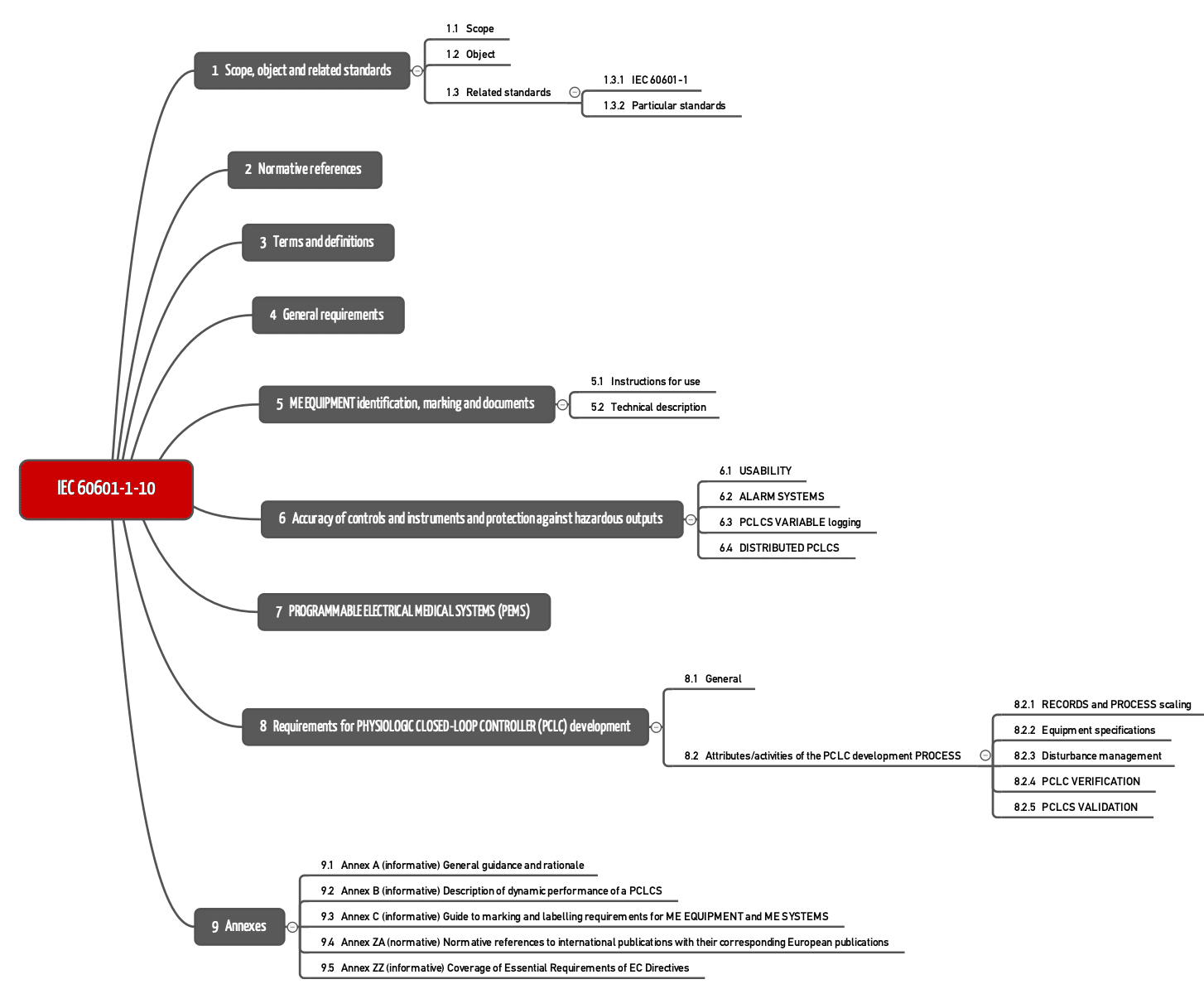
Die IEC 60601-1-10 ist eine horizontale Norm. Der Gegenstand sind Grundprinzipien, Konzepte und Begriffe. Das bedeutet, dass die Norm keine Anforderungen für eine bestimmte Art von Medizinprodukten festlegt. Solche besonderen Festlegungen befinden sich in den Part-2 Normen.
Die IEC 60601-1-10 formuliert die Anforderungen in den Kapiteln 4 bis 8.
Kapitel 4: Allgemeine Anforderungen
Das kompakte vierte Kapitel listet konkrete Parameter und Gefährdungen, die die Hersteller im Risikomanagement betrachten müssen. Dazu zählen Latenzzeiten, die unterschiedliche Reaktion verschiedener Patienten auf die Steuerung und der Einfluss der Elemente des Regelkreises, z. B. des Sensors.
Kapitel 6: Ausgangsgrößen
Die Norm stellt Anforderungen an die Outputs. Das betrifft die
- Gebrauchstauglichkeit des „User Interfaces“,
- Existenz (falls ein Fallback notwendig ist) und Konformität der Alarme (mit Hinweis auf die IEC 60601-1-8),
- Verpflichtung zum Logging (ohne auf Datenschutzaspekte einzugehen) und
- Verwendung von verteilten Regelsystemen (die die Norm explizit gestattet).
Kapitel 7: PEMS
Die Norm referenziert im Kapitel sieben lediglich das Kapitel 14 der IEC 60601-1.
Kapitel 8: Anforderungen an die Entwicklung
Das achte Kapitel stellt Anforderungen daran, was die Hersteller spezifizieren und welche risikominimierenden Maßnahmen sie berücksichtigen müssen. Beispielsweise müssen sie festlegen, wie sich das Produkt während des Normalzustands sowie im Worst-Case-Fall bei der Stellgröße, der Messgröße und dem Patienten-Transferelement verhalten soll.
Falls notwendig, sollen die Hersteller die Bereiche der Variablen einschränken. Ein Beispiel hierfür ist die maximale Wärmemenge, die einem Patienten zugeführt werden soll.
Weiterhin legt das Kapitel fest, welche Störungen und damit Gefährdungen die Hersteller analysieren und beherrschen müssen.
Schließlich fordert die Norm die Verifizierung und Validierung der Regelkreise und benennt dafür Methoden.
Die IEC 60601-1-10 war unter der MDD und AIMD harmonisiert. Die Harmonisierung unter der MDR ist noch nicht erfolgt.
Sie können die IEC 60601-1-10 beim EVS kaufen zu einem Bruchteil der Kosten, die die ISO, das IEC oder Beuth verlangen.
d) Weitere Normen
Weitere Normen wie die ISO 80601-2-13 für Anästhesiegeräte oder die IEC 60601-2-20 für Säuglingsinkubatoren gehen spezifisch auf Gerätetypen ein und stellen Anforderungen, die sich letztlich an die Regelkreise richten.
Es gibt Part-2-Normen, die die IEC 60601-1-10 explizit als nicht anwendbar betrachten. Das betrifft in der Regel Geräte, die mit Regelungen arbeiten, die aber keine physiologische Größe führen. Beispiele dafür sind CT Röntgengeräte (IEC 60601-2-44) oder Lithotripsiegeräte (IEC 60601-2-36).
Einige Normen, z. B. die DIN EN 60601-2-12 bzw. deren aktuelle Version DIN EN ISO 80601-2-12:2012-02 für die Beatmungsgeräte, gehen spezifisch auf Gerätetypen ein. Allerdings zählen die Beatmungsgeräte meist nicht zu den geschlossenen Regelkreisen:
ventilators are not considered a physiologic closed-loopclosed control system …
ISO 80601-2-12, 201.1.1
5. Typische Herausforderungen bei geschlossenen Regelsystemen
a) Entwicklung von Closed-Loop-Algorithmen
Die Regelungs- und Steuerungstechnik ist eine umfassende wissenschaftliche Disziplin, der sich ganze Studiengänge und Lehrstühle widmen. Zu den typischen Herausforderungen, vor denen die Entwickler stehen, zählen etwa die folgenden:
- Ein Regelkreis soll stabil sein: Auch bei großen Störgrößen muss das System wieder zum Führungswert zurückkehren.
- Regelkreise müssen ausreichend schnell sein: Die Zufuhr eines Medikaments (Insulin, Narkosemittel, Elektrolyte) muss eher in Sekunden als in Minuten den Sollwerten genügen.
- Der Regelkreis muss die Sollwerte ausreichend genau erreichen: Ein pH-Wert von 7,42 ist okay. Ein pH-Wert von 7,7 bedeutet den Tod des Patienten.
b) Verifizierung von Closed-Loop-Systemen
Die Sicherheit einfacher Regelkreise lässt sich auch formal beweisen. Bei hochkomplexen und voneinander abhängigen Regelkreisen ist der Beweis manchmal schwierig, manchmal wie bei deterministisch-chaotischen Systemen sogar unmöglich. Jedoch müssen Hersteller die Sicherheit, Wiederholbarkeit, Zuverlässigkeit und Leistung (wörtlich gefordert) der Produkte und damit der Regelkreise nachweisen.
Zu den Ansätzen, um diese Nachweise zu erbringen, zählen:
- Modellbasiertes Testen: Setzt voraus, dass man ein verlässliches Modell der Regelstrecke (z. B. des Patienten) hat.
- Limitierung der Stellgrößen (teilweise sogar mechanisch)
- Plausibilitätsprüfungen der Stellgrößen bzw. Regelgrößen und Intervention durch Systemkomponenten außerhalb des Regelkreises (→ funktional sichere Architekturen)
- Durchprobieren möglicher Kombinationen, insbesondere der Störgrößen und deren zeitlichen Verlauf. Diese „Brute-Force-Variante“ funktioniert aber nur bei einer endlichen Anzahl zu testender Kombinationen von Inputs.
c) Human Factors
Ein besonderes Risiko sehen die Autoren im Zusammenhang mit der Bedienung. Der Bediener muss in der Lage sein, den Status des PCLC-Systems im Einklang mit seinem mentalen Modell zu verstehen. Das mentale Modell ist die Vorstellung des Bedieners, wie das PCLC-System funktioniert und aufgebaut ist. Wenn der Bediener versteht, auf welche Weise ein PCLC-System funktioniert, ermöglicht das ihm, angemessen auf Probleme zu reagieren, die bei der Benutzung des PCLC-Systems auftreten können.
Sie sprechen von den folgenden Herausforderungen
- Mangelnde Transparenz (für Anwender)
- „Automation bias“ (blindes Vertrauen der Anwender)
- Anwender verlieren die Fähigkeit, Entscheidungen in der bisherigen Güte zu treffen („Skill degradiation“)
6. Fazit und Ausblick
Viele medizinische Behandlungen sind zu komplex oder zu zeitkritisch, um fehlerfrei von Anwendern in Gänze durchgeführt zu werden. Closed-Loop-Systeme können diese Aufgaben ganz oder teilweise übernehmen.
Es ist offensichtlich, dass der Einsatz dieser geschlossenen Regelkreise in Zukunft weiter zunehmen wird. Dazu trägt auch das Machine Learning bei, das hilft, neue Modelle des Körpers zu entwickeln und in diesen Regelkreisen zu berücksichtigen.
Die Anforderungen an die Personen, die diese Systeme entwickeln, sind ebenso hoch wie jene an die Personen, die diese Closed-Loop-Systeme verifizieren und validieren.
Regel 22 der MDR zeigt, dass die europäischen Gesetzgeber sich erste Gedanken zu diesem Thema machen. Die Definitionen und Anforderungen der Gesetzgeber bezüglich Closed-Loop-Systemen sollten präzisiert werden.
Bei der FDA liegen Interpretationshilfen bereits in Form einer Leitlinie vor. Deren Anforderungen decken sich weitestgehend mit den Anforderungen in der IEC 60601-1-10. Die FDA-Leitlinie referenziert die IEC 60601-1-10 an vielen Stellen. Hersteller sollten dennoch beide Dokumente berücksichtigen.
Lesen Sie hier mehr über die Medizinprodukteverordnung MDR und die Änderungen.
Melden Sie sich, falls Sie die Konformität Ihrer Closed-Loop-Systeme sicherstellen wollen.
Dank an DrMW für wertvolle Anregungen
Änderungshistorie
- 2025-01-13: Beispiel am Beginn des Artikels eingefügt, interne Verlinkung ergänzt, Beginn des Abschnitts 4.a) umformuliert und direkt davor Hinweis auf autonome Systeme ergänzt
- 2023-10-27: Kapitel 4.b) wegen neuer FDA Guidance komplett überarbeitet
- 2022-03-14: Artikel grundlegend überarbeitet; Abschnitte zur IEC 60601-1-10 und zum Guidance-Dokument der FDA ergänzt


Lieber Herr Johner,
erwähnenswert wäre in diesem Kontext sicher auch die IEC 60601-1-10 „Anforderungen an die Entwicklung von physiologischen geschlossenen Regelkreisen“, die neben der generellen Betrachtung solcher Regelkreise auch Aspekte wie Usability, Alarmsysteme und Logging einschließt. Anforderungen an die Validierung solcher Systeme finden sich ebenfalls – obwohl mir die Möglichkeit einer Validierung alleine durch Literaturstudien etwas schmal erscheint.
Beste Grüße,
O. Thilmann
Sie haben uneingeschränkt Recht, lieber Herr Thilmann: Die IEC 60601-1-10 verdient der Erwähnung. Dies ist mit einem weiteren Beitrag auch geplant.
Danke für Ihren Hinweis, mit dem dieser Artikel bereichert ist.
Sehr geehrter Herr Professor Johner,
vielen Dank für Ihre Erläuterungen.
Ich stimme Ihnen zu, dass die Regel 22 sehr problematisch ist, in dem sie nach dem Wortlaut im Beispiel alle technischen Regelkreise einschließt und nicht auf die engere Definition der IEC 60601-1-10:2007 abhebt, das eine physiologische Variable als Patiententransferelement voraussetzt (3.18, 3.19, 3.20).
Einen AID würde ich schon wegen seiner „diagnostischer Funktion“ unter der Regel 22 sehen, auch ohne geschlossenen Regelkreis. Nach meinem Verständnis bezieht Regel 22 aber beide Beispiele als Arten von Medizinprodukten ein.
Primär hebt Regel 22 auf „diagnostische Funktion“ und „erheblichen Einfluss auf das Patientenmanagement ab“, und nennt dann zwei nur bedingt passende Beispiele.
Mit freundlichen Grüßen, Jens-Uwe Hagenah
Danke für Ihre Gedanken, denen ich absolut zustimme.
Lieber Herr Johner,
vielen Dank für den übersichtlichen Artikel zu den Closed Loop Systemen.
Bezüglich der MDR Regel 22 möchte ich folgende Gedanken beisteuern und halte eine differenziertere Betrachtung für notwendig:
Die Regel 22 hat zwei Teile: a) die Anforderung „Active therapeutic devices with an integrated or incorporated diagnostic function which significantly determines the patient management by the device“ und b) den Teil Beispiel „such as closed loop system or automated external defibrillators, are classified as class III.“
Der Anforderungsteil stellt erstmal keinen Bezug zu Closed Loop Systemen her.
Der Anfoderungsteil besteht auch wieder aus zwei Anforderungen: a) das Produkt muss eine „integrated or incoprorated diagnostic function“ besitzen. Diagnostic Function ist leider nihct weiter spezifiziert. ISt eien reine Messung und Anzeige eines Messwerts schon eine „diagnostic function“? Oder erst wenn das Produkt zu dem Ergebnis kommt z.B. „Blutzuckerspiegel ist zu hoch / zu niedrig“ ? Oder das Produkt erkennt eigenständig eine bestimmte Krnakheit, z.B. Sepsis? Hierzu hätte ich mir auch in der MDCG zur Klassifizierung mehr Interpreation gewünscht.
Dann kommt der 2. Teil der Anforderung – der genauso Interpretationswürdig ist: „which significantly determines the patient management“. Was bedeutet das nun genau? Ein Regelkreis (egal ob nun physiologisch oder technisch) der nur den vom Arzt eingestellten Zielwert einhält und somit etwaige Störungen ausgleicht, würde ich z.B. nihct als signifikant fürs Patient Management sehen, denn es wird ja nur der vom Arzt vorgegebene Wert einghalten. Die korrekte und angemessene Wahl des Soll- und Zielwertes durch den Arzt selber sehe ich viel signifikanter für das poatient management.
Wann wäre denn etwas signifikant für das Patient Management? Doch erst wenn durch eine „Diagnose“ das System automatisch auch die Therapie einleitet und auch die therapierelevanten Parameter und Sollwerte automatisiert festlegt werden.
Somit wäre zumindest recht eindeutig, dass für die allermeisten Closed Loop Systeme die Regel 22 nicht relevant wäre.
Warum die Autoren der Regel 22 als Beispiele „closed loop systems“ erwähnt haben, bleibt unklar und wird leider auch durch die MDCG zur Klassifizierung nicht verständlicher. Aus den oben dargelegeten Überlegeungen müssten es schon sehr spezielle Closed Loop Systeme sein, eher schon „autonome“ Systeme.
Mit freundlichen Grüßen
Stefan Meyer
Lieber Herr Meyer,
Vielen Dank für das Teilen Ihrer Ansichten – ich stimme Ihnen zu. Ich glaube nicht, dass die MDR bei den Beispielen in Regel 22 speziell die IEC 60601-1-10 berücksichtigt hat.
Daher würde ich nicht daraus ableiten, dass alle ME-Geräte, die unter den Geltungsbereich der IEC 60601-1-10 fallen, automatisch unter Regel 22 eingeordnet und als Klasse-III-Produkte klassifiziert werden müssen. Die Anwendbarkeit von Regel 22 erfordert eine differenzierte Bewertung anhand Ihres Schemas – jedoch unabhängig von der Definition der IEC 60601-1-10. Meiner Interpretation nach zielt Regel 22 darauf ab, Systeme mit einer bestimmten Regelungstiefe höher einzustufen, insbesondere wenn der Anwender die Regelung nicht vollständig nachvollziehen kann und dadurch ein erhöhtes Risiko entsteht.
Herzliche Grüsse, Mario Klessascheck