
Der Einsatz von CMR-Stoffen ist streng reguliert. Auch die MDR regelt die CMR-Stoffe und stellt strenge Anforderungen an die Medizinproduktehersteller.
Dieser Artikel hilft, diese Anforderungen zu erfüllen.
1. CMR-Stoffe: Die Herausforderungen
CMR steht für:
- Cancerogen (kanzerogen, krebserzeugend)
- Mutagen (erbgutverändernd)
- Reproduktionstoxisch (fortpflanzungstoxisch)
Für Medizinproduktehersteller sind die CMR-Stoffe herausfordernd:
- CMR-Stoffe haben besonders negative Auswirkungen auf die Patientensicherheit.
- Die Folgen für die Patienten sind jedoch nur langfristig erkennbar und meist nicht über die Post-Market Surveillance abdeckbar.
- Es ist nicht sofort klar, welche Stoffe als CMR (kanzerogen, mutagen oder reproduktionstoxisch) gelten.
- Die gesetzlichen Anforderungen sind (deshalb) besonders umfangreich.
- Ein Nachweis der Abwesenheit von CMR-Stoffen und damit der Konformität ist schwierig.
Die folgenden Kapitel helfen dabei, diese Herausforderungen zu bewältigen.
2. Einstufung als CMR-Stoff
Hersteller müssen wissen, welche Stoffe ihre Produkte enthalten und freisetzen. Sie müssen auch wissen, ob diese Stoffe als kanzerogen, mutagen oder reproduktionstoxisch gelten.
Bei dieser Einteilung helfen Datenbanken.
a. Toxikologische Datenbanken
Toxikologische Datenbanken enthalten Daten zu In-vitro– und In-vivo-Versuchen, die Aufschluss darüber geben, ob eine Substanz kanzerogen, mutagen oder reproduktionstoxisch wirkt.
Beispiele für toxikologische Datenbanken sind:
Die QSAR-Toolbox kann verschiedene Datenbanken abfragen.
b. Anhang der CLP-Verordnung
Die CLP-Verordnung enthält in der Tabelle 3 des Anhangs VI eine Liste von CMR-Stoffen mit ihrer offiziellen Einstufung.
Der offizielle Titel der Verordnung lautet „Verordnung (EG) Nr. 1272/2008 des Europäischen Parlaments und des Rates vom 16. Dezember 2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen“.
Bei dieser Einstufung helfen Datenbanken. Beachten Sie hierbei, dass diese Einstufung in den Datenbanken oftmals auf unterschiedlichen Studiendaten beruht und sich daher unterschiedliche Schlussfolgerungen/Klassifizierungen ergeben können.
Bei sich widersprechenden Daten hilft die Einschätzung durch einen Fachexperten mit toxikologischer Kenntnis.
c. Liste der EU-Kommission
Die EU hat eine Übersicht zum Thema CMR-Substanzen veröffentlicht, die allerdings einen Fokus auf Kosmetika legt. Diese Stoffe können jedoch auch in Medizinprodukten gefunden werden und sind daher relevant.
d. KMR-Liste des IFA
Das Institut für Arbeitsschutz der Deutschen Gesetzlichen Unfallversicherung (IFA) hat eine KMR-Liste publiziert. Diese umfassende Liste enthält als CMR eingestufte Stoffe und wird regelmäßig aktualisiert.
e. IARC-Monografie
Auf der Webseite der EU findet sich eine Liste der IARC (International Agency for Research on Cancer).
3. Herkunft der CMR-Stoffe in Medizinprodukten
Es gibt viele verschiedene Möglichkeiten, wie CMR-Substanzen in Produkte eingebracht werden können. Nicht immer lässt sich der Ursprung eindeutig klären. Möglichkeiten sind:
- Bereits im Ausgangsmaterial enthaltene CMR-Stoffe
- Die Stoffe sind Rückstände von Prozesschemikalien.
- Die CMR-Stoffe entstehen bei der Produktion, z. B. beim Erhitzen oder durch Reaktionen mit anderen Materialien.
4. Nachweis von CMR-Stoffen
Der Nachweis und die Bewertung der CMR-Stoffe erfolgen in mehreren Schritten.
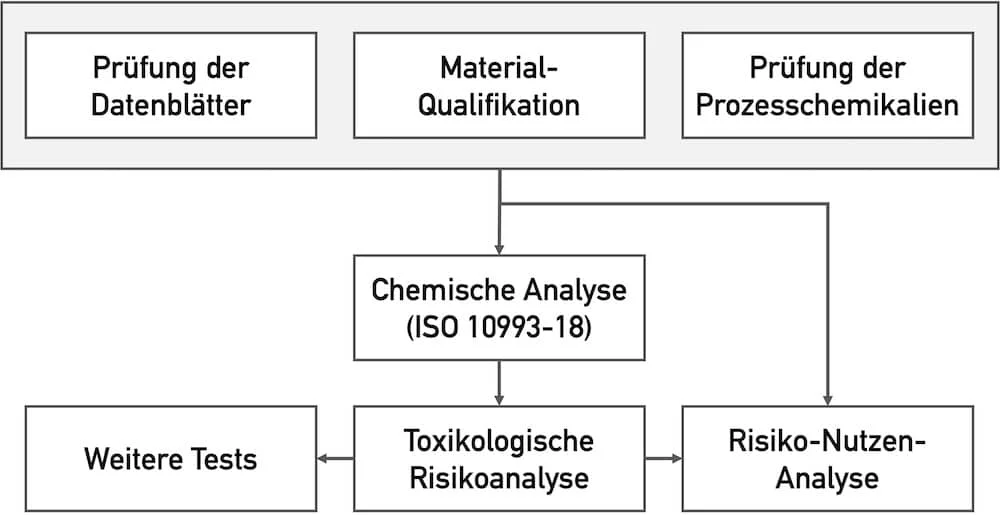
Schritt 1: Datenblätter prüfen
Im ersten Schritt sollten die Hersteller die Datenblätter der Ausgangsmaterialien prüfen und die Eignung des Materials im Zuge einer Materialqualifikation sicherstellen. Das Gleiche gilt für die Datenblätter der Prozesschemikalien.
Schritt 2: Chemische Analyse durchführen
Ob das finale Produkt CMR-Stoffe freisetzt, klärt letztlich die chemische Analyse. Diese sollte zur chemischen Charakterisierung des Produkts gemäß ISO 10993-18 immer ein Teil der für die Biokompatibilität notwendigen Prüfungen sein. Die Ergebnisse sollten von einem Experten mit toxikologischer Fachkenntnis bewertet werden.
Schritt 3: Toxikologische Risikoanalyse & weitere Tests
Auf Grundlage der chemischen Charakterisierung kann eine toxikologische Bewertung erfolgen. Diese klärt im Abgleich mit relevanten Grenzwerten, wie das Risiko durch eine mögliche Freisetzung von CMR-Substanzen einzuschätzen ist und ob weitere Tests notwendig sind (z. B. Prüfungen auf Gentoxizität, Karzinogenität und Reproduktionstoxizität gemäß ISO 10993-3).
5. Regulatorische Anforderungen
a. Anforderungen der MDR
MDR, Kapitel II, 10.4.1
Produkte, die invasiv sind oder direkt mit dem Körper interagieren, um Medikamente oder Körperflüssigkeiten zu verabreichen, zu entnehmen oder zu lagern, dürfen CMR-Stoffe in einer Konzentration über 0,1 % Massenanteil nur enthalten, wenn es gerechtfertigt ist.
MDR, Kapitel II, 10.4.2
Die Rechtfertigung von CMR-Stoffen muss enthalten:
- Eine Analyse der potenziellen Exposition
- Die Untersuchung verfügbarer Alternativen, einschließlich wissenschaftlicher Bewertungen und Studien
- Eine Begründung, warum unter Berücksichtigung der Produktfunktionalität und des Nutzen-Risiko-Verhältnisses Alternativen unangemessen sein könnten
- Die Berücksichtigung spezieller Patientengruppen (Kinder, Schwangere, Stillende).
Falls vorhanden, sollten aktuelle Leitlinien wissenschaftlicher Ausschüsse herangezogen werden.
b. ISO 10993-Familie
Im Rahmen der Bewertung der biologischen Sicherheit nach ISO 10993-1 sind die Endpunkte Genotoxizität (schließt u. a. mutagenes Potenzial von Substanzen ein), Kanzerogenität, Reproduktions- und Entwicklungstoxizität zu adressieren.
Im Rahmen der toxikologischen Risikobewertung gibt die 10993-17 Auskunft über die Risikobewertung von kanzerogenen Substanzen. Diese Bewertungen sollten immer von Experten mit entsprechender toxikologischer Fachkenntnis durchgeführt werden.
c. Weitere Anforderungen an CMR-Stoffe
Indirekt wirken sich weitere Regularien aus: Die REACH-Verordnung regelt den Umgang mit besonders besorgniserregenden Stoffen (SVHC), zu denen viele CMR-Stoffe zählen. Die CLP-Verordnung regelt die Einstufung, Kennzeichnung und Verpackung von chemischen Stoffen. Außerdem sind die nationalen Regularien zu beachten (z. B. die Gesetzgebung zum Arbeitsschutz und die Gefahrstoffverordnung).
Sofern Sie CMR-Stoffe in Ihrem Produkt nicht vermeiden können, achten Sie auf die Anpassung der Dokumentation (Label, IFU).
Die gesetzlichen Vorgaben dazu beschreibt der Fachartikel zum Labeling bei Medizinprodukten.
6. Tipps zum Erfüllen der Anforderungen an CMR-Stoffe
Tipp 1: Von Anfang an auf CMR-freie Stoffe achten
Verwenden Sie nur Ausgangsstoffe, für die Sie aussagefähige Datenblätter erhalten. Prüfen und bewerten Sie die darin genannten Substanzen und vermeiden Sie Ausgangsmaterialien, die CMR-Stoffe sind oder enthalten.
Führen Sie auf dieser Grundlage eine Materialqualifikation durch und dokumentieren Sie, warum das Material geeignet ist. Ziehen Sie schon an dieser Stelle Fachexperten zurate.
Wählen Sie bei allen Schritten der Produktion nur Materialien, die frei von CMR-Stoffen sind.
Falls die Substanzen nicht vermeidbar sind, dann streben Sie an, dass deren Konzentration unterhalb der Grenze von 0,1 % Massenanteil bleibt. Falls Sie diese Grenze nicht einhalten können, lassen Sie eine Expositionsabschätzung und eine Risiko-Nutzen-Analyse von einem Experten mit toxikologischer Fachkenntnis durchführen.
Tipp 2: Die Prüfungen früh im Projekt durchführen
Zu oft stellen Medizinproduktehersteller das Thema CMR „hinten an“ und führen die notwendigen Untersuchungen erst am Ende der Produktentwicklung durch. Das schränkt die Handlungsoptionen ein und bringt oft hohe Nachbesserungskosten und Projektverzug mit sich.
Eine frühe Kenntnis der Stoffe ermöglicht oft ein Re-Design des Produkts, um die Freisetzung von CMR-Substanzen aus dem finalen Produkt zu vermeiden bzw. zu reduzieren.
Tipp 3: Die ISO 10993-Familie befolgen
Prüfen Sie, wie von der ISO 10993 vorgegeben, die Biokompatibilität am finalen Produkt. Dazu zählt zwingend auch eine ausführliche chemische Charakterisierung.
Bewerten Sie die Ergebnisse und führen Sie bei Bedarf eine toxikologische Risikobewertung durch.
Tipp 4: Die Expertise sicherstellen
Achten Sie auf die dokumentierte Expertise der dafür verantwortlichen Personen. Diese müssen auch toxikologische Details kennen und z. B. die Exposition und die Auswirkungen auf kritische Patientengruppen bewerten können.
7. Fazit und Zusammenfassung
Dass die Gesetzgeber besonders strenge Anforderungen an kanzerogene, mutagene und reproduktionstoxische Stoffe stellen, ist nachvollziehbar.
Daher sollten die Hersteller diese Substanzen bestmöglich in ihren Medizinprodukten vermeiden und die Nachweise führen, dass die CMR-Stoffe unterhalb der toxikologisch anwendbaren Grenzwerte bleiben. Eine toxikologische Risikobewertung ist dennoch notwendig und stellt hohe Kompetenzanforderungen an die dafür Verantwortlichen.
Das Johner Institut hilft Herstellern von Medizinprodukten bei dem Nachweis, dass alle gesetzlichen Anforderungen (auch) an die CMR-Stoffe erfüllt sind, und damit bei der schnellen Zulassung sicherer Medizinprodukte.
- Die Experts des Johner Instituts prüfen pragmatisch die biologische Sicherheit, führen chemische Charakterisierungen durch und bewerten die Ergebnisse.
- Sie helfen auch bei der Bewertung und Einordnung von bereits vorhandenen Prüfergebnissen im Hinblick auf die aufgefundenen CMR-Stoffe.
- Bei Produkten, die CMR-Stoffe enthalten, helfen unsere Experts mit Risiko-Nutzen-Analysen.
- Das Johner Institut bietet flexible Beratung bei allen Fragen, allen Produkten und allen Märkten.
Melden Sie sich, z. B. über unsere Kontaktseite, und Sie erhalten sofort Unterstützung.



Guten Tag Frau Domke,
vielen Dank für Ihren Artikel. CMR-Stoffe sollten grundsätzlich in Medizinprodukten vermieden werden. Warum gibt die MDR dann einen Grenzwert von 0.1% Massenanteil vor? Bedeutet dieses, dass ein Massenanteil von kleiner als 0.1% akzeptierbar ist?
Freundliche Grüsse,
Dirk Steinhoff
Lieber Herr Steinhoff,
vielen Dank für Ihre Nachfrage.
Der in der MDR angegebene einen Grenzwert von 0,1 % Massenanteil für CMR-Stoffe ist nach meiner Einschätzung so festgelegt, um einen machbaren Kompromiss zwischen Sicherheit und Funktionalität zu erreichen.
Dabei ist wichtig, dass der Grenzwert nicht bedeutet, dass ein Massenanteil von weniger als 0,1 % automatisch akzeptabel ist. Vielmehr dient er als Schwellenwert, ab dem besondere Rechtfertigungen und Maßnahmen erforderlich sind, wie etwa eine Risiko-Nutzen-Abwägung.
Die Konzentration sollte immer so niedrig wie möglich gehalten werden, auch wenn sie unter 0,1 % liegt.
Beste Grüße
Tanja Domke