
Companion Diagnostics (auch CDx oder therapiebegleitende Diagnostika) kommen gemeinsam mit einem Arzneimittel zum Einsatz. Mit ihnen können Ärztinnen und Ärzte beispielsweise sicherstellen, ob ein bestimmtes Arzneimittel für den Patienten tatsächlich geeignet ist. CDx spielen daher eine besonders große Rolle in der personalisierten Medizin.
Da das therapiebegleitende Diagnostikum und das Arzneimittel untrennbar miteinander verbunden sind, gelten bei Entwicklung und Zulassung einige Besonderheiten.
In diesem Beitrag erfahren Sie,
- wie Companion Diagnostics definiert werden,
- wie die Entwicklung und Zulassung von CDx in der Praxis aussieht
- welche regulatorischen Hürden sich dabei in Europa und den USA ergeben
- welche Tipps wir für Sie haben, damit die Entwicklung und Zulassung von CDx gelingen.
1. Was sind Companion Diagnostics (CDx)?
Der Erfolg einiger medikamentöser Therapien setzt den Einsatz eines In-vitro-Diagnostikums (IVD) voraus. Mit diesem IVD stellen Labore beispielsweise fest, ob ein Biomarker, an dem das Medikament ansetzt, beim Patienten überhaupt vorhanden ist. Mit der IVD-Analyse können Ärztinnen und Ärzte sicherstellen, dass eine bestimmte Therapie für den jeweiligen Patienten tatsächlich geeignet ist.
Üblicherweise weist das CDx in den jeweiligen Analyten genetische Sequenzveränderungen oder das Vorhandensein bestimmter Proteine nach.
Typische Plattformen sind:
- Immunhistochemie
- Quantitative PCR
- Next-Generation-Sequencing
- In-situ-Hybridisierung (FISH, CISH)
Ist das Ergebnis des IVDs die Voraussetzung für eine sichere Arzneimitteltherapie, spricht man von einem therapiebegleitenden Diagnostikum (Companion Diagnostics, CDx). CDx werden im Idealfall in enger Zusammenarbeit mit Arzneimittelherstellern entwickelt.
Aus dem Erwägungsgrund (11) der IVDR ergeben sich vor allem die folgenden Anwendungszwecke von CDx:
- durch quantitative oder qualitative Bestimmung spezifischer Marker die Eignung von Patient:innen für eine bestimmte Behandlung mit einem Arzneimittel feststellen
- Patient:innen identifizieren, bei denen ein erhöhtes Risiko einer unerwünschten Reaktion auf das betreffende Arzneimittel besteht
- Patient:innen identifizieren, für die das therapeutische Erzeugnis angemessen untersucht und als sicher und wirksam befunden wurde. Solche Biomarker können bei gesunden Personen und/oder bei Patient:innen vorliegen.
Allerdings unterscheiden sich die Definitionen von CDx in Europa und den USA.
a) Unterschiedliche Definitionen von CDx
Europa
In Europa versteht die Verordnung 2017/746 über In-vitro-Diagnostika (IVDR) unter “therapiebegleitendem Diagnostikum”:
„therapiebegleitendes Diagnostikum“ bezeichnet ein Produkt, das für die sichere und wirksame Verwendung eines dazugehörigen Arzneimittels wesentlich ist, um
- a) Patienten vor und/oder während der Behandlung zu identifizieren, die mit der größten Wahrscheinlichkeit von dem dazugehörigen Arzneimittel profitieren, oder
- b) Patienten vor und/oder während der Behandlung zu identifizieren, bei denen wahrscheinlich ein erhöhtes Risiko von schwerwiegenden unerwünschten Reaktionen infolge einer Behandlung mit dem dazugehörigen Arzneimittel besteht;
Quelle: Art. 2 (7) IVDR
Explizit nicht als CDx gelten:
“Produkte, die zur Überwachung der Behandlung mit einem Arzneimittel eingesetzt werden, um sicherzustellen, dass die Konzentration der betreffenden Stoffe im menschlichen Körper innerhalb des Therapiefensters liegt”.
Erwägungsgrund (12) IVDR
Diese Ausnahme ist besonders auch deshalb wichtig, da es sie in den USA nicht gibt. Die Definition der FDA zu therapiebegleitenden Diagnostika unterscheidet sich von der europäischen:
USA
Die FDA definiert “IVD companion diagnostic devices” wie folgt:
„An IVD companion diagnostic device could be essential for the safe and effective use of a corresponding therapeutic product to:
- Identify patients who are most likely to benefit from the therapeutic product
- Identify patients likely to be at increased risk for serious adverse reactions as a result of treatment with the therapeutic product
- Monitor response to treatment with the therapeutic product for the purpose of adjusting treatment (e.g., schedule, dose, discontinuation) to achieve improved safety or effectiveness
- Identify patients in the population for whom the therapeutic product has been adequately studied, and found safe and effective, i.e., there is insufficient information about the safety and effectiveness of the therapeutic product in any other population”
Quelle: FDA
Hersteller, die ihr In-vitro-Diagnostikum zur Überwachung einer Behandlung mit einem Arzneimittel sowohl im europäischen als auch im US-amerikanischen Markt in Verkehr bringen wollen, sollten diese unterschiedlichen Definitionen unbedingt beachten.
b) Abgrenzung zu Complementary Diagnostics
Es existiert eine weitere Gruppe von IVD, die zwar keine therapiebegleitenden Diagnostika (Companion Diagnostics) sind, aber mit ihnen verwechselt werden könnten: die sogenannten Complementary Diagnostics (frei übersetzt “ergänzenden Diagnostika”).
Diese ergänzenden Diagnostika werden in der IVDR weder definiert noch beschrieben. Sie werden im Zusammenhang mit einem bestimmten Arzneimittel eher empfohlen, als dass sie verbindlich vor der Anwendung des Arzneimittels einzusetzen sind. Es handelt sich daher lediglich um eine Entscheidungshilfe, nicht um einen notwendigen Bestandteil der Therapie.
Die enge Verknüpfung mit einem Medikament, wie sie bei therapiebegleitenden Diagnostika (CDx) vorliegt, gibt es demnach bei therapieergänzenden Diagnostika nicht. Complementary Diagnostics werden daher regulatorisch wie “herkömmliche IVD” behandelt, d. h., dass die Besonderheiten, die sich für CDx ergeben, keine Anwendung finden.
Beispiel:
Ein von der FDA zugelassenes Complementary Diagnostic ist ein immunhistochemischer Assay zum Nachweis des PD-L1-Proteins in Patient:innen, die an nicht-kleinzelligem Lungenkarzinom leiden und für eine Krebsimmuntherapie (Nivolumab) in Betracht kommen.
Es handelt sich um ein Complementary Diagnostic und kein CDx, da ein therapeutischer Nutzen für ALLE Patient:innen, die das Arzneimittel erhielten, nachgewiesen werden konnte, unabhängig vom Status des Biomarkers. Dennoch gibt es einige Patient:innen, die besser auf das Arzneimittel anschlagen als andere: Das sind diejenigen, die besonders hohe Level des Proteins PD-L1 exprimieren.
Der Einsatz des IVD ist demnach nicht zwingend erforderlich, gibt jedoch eine Prognose, wie wirksam die Arzneimittelbehandlung sein könnte.
- Guidance Dokument MDCG 2020-16 rev.2 mit Flussdiagramm zur Bestimmung als CDx in Anhang II
- Glossar der relevanten Begriffe zu Companion Diagnostics (CDx) und Complementary Diagnostics
- Veröffentlichung Clinical Evidence Requirements for CE certification under the In Vitro Diagnostic Regulation in the European Union von MedTech Europe
2. CDx in der Praxis
a) Für wen CDx relevant sind
- Arzneimittelhersteller
- Arzneitmittelhersteller sollten sich mit den regulatorischen Basics für IVD und den Abhängigkeiten von IVD und Arzneimittel auseinandersetzen.
- Arzneitmittelhersteller sollten sich mit den regulatorischen Basics für IVD und den Abhängigkeiten von IVD und Arzneimittel auseinandersetzen.
- IVD-Hersteller
- IVD-Hersteller, die die Entwicklung von CDx erwägen oder bereits durchführen, sollten sich mit den Besonderheiten in Entwicklung und Zulassung vertraut machen. Für CDx bestehen höhere regulatorische Hürden als für andere IVD derselben Risikoklasse.
b) Einsatzgebiete von CDx
CDx spielen eine große Rolle in der “personalisierten Medizin”. Hierbei geht es darum, eine maßgeschneiderte Therapie für den individuellen Patienten bzw. die Ursachen seiner Erkrankung festzulegen und ggf. fortlaufend anzupassen. Klassischerweise finden CDx im Bereich onkologischer Fragestellungen Anwendung. Das zeigt auch ein Blick auf die List of Cleared or Approved Companion Diagnostic Devices der FDA.
Eines der ersten CDx war ein Nachweissystem zur Bestimmung des Expressionslevels von HER-2 (auch bekannt als ERBB2) in Brustkrebsgewebe. Dieser Test basiert auf einem immunhistochemischen Verfahren und dient der Bestimmung, ob Patient:innen für die Behandlung mit Herceptin (Trastazumab) geeignet sind. Herceptin wiederum ist für die Behandlung von metastatischem Brustkrebs zugelassen. Dem Arzneimittel fehlt die Wirksamkeit in HER-2-negativen Patient:innen und kann im Weiteren sogar negative Auswirkungen haben. Daher ist der Einsatz eines CDx essenziell, um Patient:innen zu identifizieren, die von der Therapie profitieren können.
c) Wie die Co-Entwicklung von CDx abläuft
Da CDx und Arzneimittel voneinander abhängig sind, verläuft die Entwicklung (und Zulassung) parallel. Es ergeben sich verschiedene Phasen der Entwicklung von CDx, die mit den klinischen Phasen des Arzneimittels korrespondieren. Abbildung 1 gibt einen Überblick. Weiter unten erläutern wir die einzelnen Phasen genauer.
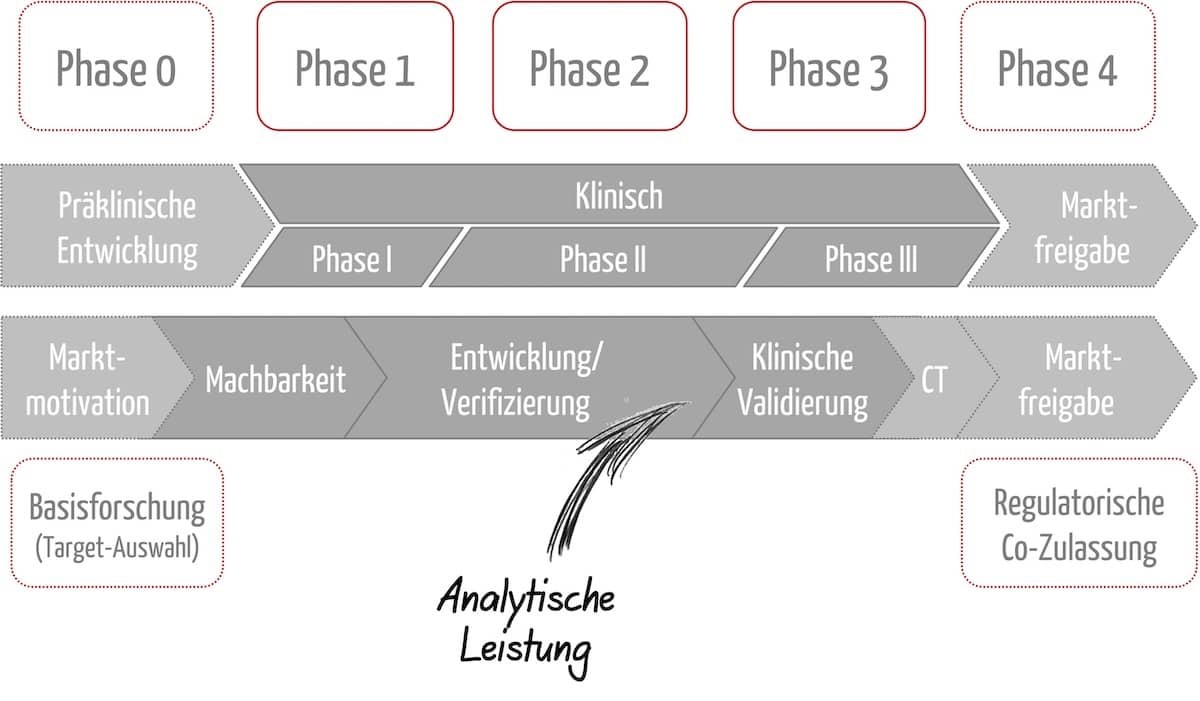
Phase 0:
- Arzneimittel
- Präklinische Phase
- Klären der Fragen: Welche Art von Diagnostik wird benötigt, um die Patientenpopulation korrekt für das Arzneimittel zu identifizieren bzw. um das Krankheitsgeschehen des individuellen Patienten zu verstehen?
- IVD (CDx)
- Grundlegende Forschung/Entwicklung
- u. a. Auswahl von Analyt(en) und Methode. Hierbei sollte der IVD-Hersteller die wissenschaftliche Validität nachweisen, die den Zusammenhang zwischen dem Analyten und dem klinischem Zustand / der Erkrankung herstellt. Dazu sollte er wissenschaftliche Rationalen für die Nutzung des Biomarkers liefern.
Phasen 1 bis 3
- Arzneimittel
- Klinische Phasen I–II
- Planung und Durchführung der Phase-III-Studie unter Einbindung des CDx
- IVD (CDx)
- Während das Arzneimittel in die klinische Phase I und II eintritt, durchläuft das CDx die gelenkte Produktentwicklung und die analytische Leistungsbewertung.
- Die analytische Leistungsbewertung, d. h. : die Fähigkeit eines Produkts, einen bestimmten Analyten korrekt nachzuweisen oder zu messen, sollte unbedingt abgeschlossen sein, bevor die Phase-III-Studie des Arzneimittels initiiert wird, um eine sinnvolle Studienplanung zu gewährleisten.
- Anschließend folgt die klinische Leistungsbewertung des IVD und die klinische Prüfung des Arzneimittels. Dabei handelt es sich um zwei Studien, mit eigenen Studienprotokollen und Endpunkten, die parallelisiert und aufeinander abgestimmt ablaufen können. Die klinische Leistungsbewertung dient dem Nachweis, dass das IVD dazu geeignet ist, Patient:innen für die weitere Arzneimitteltherapie zu stratifizieren bzw. zu selektieren. Der IVD-Hersteller belegt also die Zweckbestimmung des IVD in der tatsächlichen Patientenpopulation anhand von Daten. Die klinische Studie für das Arzneimittel soll u. a. zeigen, dass die durch das IVD bestimmte Patientenpopulation von der Therapie profitiert.
Phase 4
- Arzneimittel
- Zulassung
- IVD (CDx)
- Konformitätserklärung
- für eine gleichzeitige Zulassung des Arzneimittels und die Zertifizierung des IVD besteht keine gesetzliche Vorgabe
3. Regulatorische Besonderheiten von CDx
Da der Zweck des Diagnostikums und des Medikaments untrennbar miteinander verbunden sind, greifen die regulatorischen Anforderungen an Arzneimittel und CDx ineinander. Die speziellen regulatorische Anforderungen an CDx berücksichtigen also sowohl das IVD (CDx) als auch das Arzneimittel.
a) Regulatorische Besonderheiten in Europa
Die IVDR sieht für CDx eigene Regelungen vor. Dabei stehen die folgenden Besonderheiten im Fokus:
- Die regulatorischen Anforderungen an CDx sind strikter als die an andere IVD derselben Risikoklasse
- CDx gehören der Risikoklasse C an
- Benannte Stelle und Arzneimittelbehörde arbeiten in einem Konsultationsverfahren im Rahmen der Konformitätsbewertung zusammen
- Der klinische Nachweis entspricht dem Vorgehen bei anderen IVD: wissenschaftliche Validität, analytische Leistung, klinische Leistung. Für letztere benötigt der Hersteller allerdings Studien, die aufzeigen, dass das CDx anhand eines bestimmten Biomarkers in der Lage ist, Patient:innen für eine bestimmte Therapie zu stratifizieren oder zu identifizieren.
Im Detail sehen die regulatorischen Besonderheiten folgendermaßen aus:
Risikoklasse
Companion Diagnostics fallen bei der Risikoklassifizierung nach Anhang VIII der IVDR in die Risikoklasse C. Allerdings gelten für CDx zusätzliche regulatorischen Anforderungen, die über die Anforderungen an andere Klasse C-IVD hinausgehen (s. u.). Beispiele gibt es im MDCG-Dokument 2020-16.
Besonderes Konformitätsbewertungsverfahren
Normalerweise erfolgt die Konformitätsbewertung für Klasse-C-Produkte nur für ein repräsentatives Produkt pro generischer Produktgruppe. Bei CDx ist das jedoch anders: Art. 48 (7) IVDR besagt, dass jedes einzelne CDx-Produkt das Verfahren durchlaufen muss. Damit können CDx-Hersteller die Vorteile der Produktgruppen nicht nutzen.
Konsultation der Arzneimittelbehörde
CDx können nach Anhang IX der IVDR konformitätsbewertet werden. Hierbei ist jedoch Abschnitt 5.2 des Anhangs zusätzlich zu berücksichtigen.
Besonders bemerkenswert ist, dass zusätzlich zur Benannten Stelle in zweiter Instanz eine Arzneimittelbehörde involviert wird. Das ist in der Regel die europäische Arzneimittelbehörde EMA.
- Rolle der Arzneimittelbehörde
Anhang IX Abschnitt 5.2 beschreibt die Anforderungen an die Beteiligung einer Arzneimittelbehörde als Teil des Konformitätsbewertungsverfahrens. Die Arzneimittelbehörde kann demnach entweder eine zuständige nationale Behörde oder die EMA sein. Neue Arzneimittel werden jedoch in der Regel von der EMA geprüft.
Nationale Behörden können ggf. in einigen Fällen bei der Zulassung einiger Bestandsprodukte beteiligt sein.
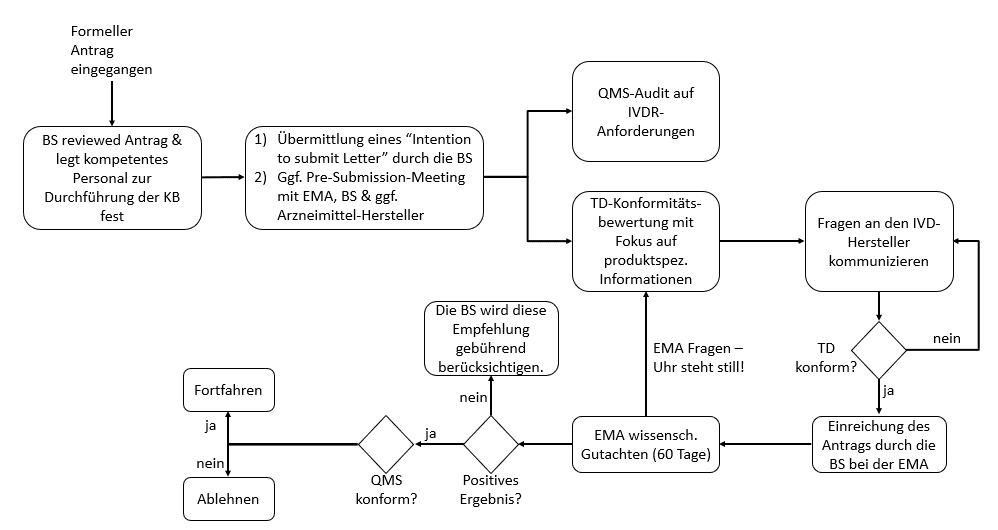
- Ablauf der Involvierung der Arzneimittelbehörde
- Die Benannte Stelle informiert die EMA über den Beginn eines Bewertungsverfahrens eines CDx, dies beinhaltet die Übermittlung einer “Absichtserklärung” (Intention to submit Letter) mind. drei Monate vor der geplanten Einreichung. (Die Inhalte des Intention to submit Letter finden Sie hier Guidance on the procedural aspects for the consultation to the European Medicines Agency by a notified body on companion diagnostics.)
- Die EMA legt den “Rapporteur” fest, diese:r leitet die Bewertung eines Antrags und ist ebenfalls für die Bewertung des zugehörigen Arzneimittel verantwortlich.
- Unterstützend kann die Benannte Stelle der EMA vor der geplanten Einreichung des Antrags Fragen zusenden. Sollte zusätzliche Anleitung erforderlich sein, kann die Benannte Stelle ein Pre-submission Meeting anfragen.
- Die Benannte Stelle ersucht ein wissenschaftliches Gutachten von der Arzneimittelbehörde hinsichtlich der Eignung des CDx in Verbindung mit dem Arzneimittel. Das Antragsformular kann auf der EMA-Website heruntergeladen werden. Antragsteller ist die Benannte Stelle
- Das wissenschaftliche Gutachten basiert auf dem Entwurf der Gebrauchsanweisung und des Kurzberichts über Sicherheit und Leistung. Bei der Bewertung hinsichtlich der Eignung des CDx in Verbindung mit dem Arzneimittel werden die wissenschaftlichen Rationale der Biomarkerauswahl, die analytische und klinische Perfomance, die klinische Sicherheit und der klinische Nutzen für die Patienten berücksichtig.
- Der Zeitrahmen für die Konsultation beträgt 60 Tage mit der Möglichkeit einer Verlängerung um weitere 60 Tage
- Bitte beachten Sie: Wenn es Rückfragen durch die Behörde gibt, wird die Frist um die Zeit verlängert, die zur Bearbeitung der Fragen benötigt wird.
- Neben diesem initialen Konsultationsverfahren muss die Behörde ggf. auch bei Änderungen des CDx einbezogen werden. Dies ist beispielsweise der Fall, wenn die Änderungen die Performance und/oder die Zweckbestimmung und/oder die Eignung des CDx in Verbindung mit dem Arzneimittel betreffen. Dann wird ein Follow-up-Konsultationsverfahren initiiert. In diesem Fall stellt die EMA ihr Gutachten innerhalb von 30 Tagen nach Erhalt aller notwendigen Dokumente zur Verfügung.
CDx basierend auf genetischen Tests: Keine Patientenberatung
Wenn ein CDx auf genetischen Tests basiert, kann die Patientenberatung, die bei anderen Gentests gefordert ist, entfallen (Art. 4 (3) IVDR).
Leistungsbewertung
Generell gilt: CDx benötigen stets klinische Daten und fallen unter “bestimmte Leistungsstudien” gemäß Art. 58 (2) IVDR.
Die drei Säulen des klinischen Nachweises – wissenschaftliche Validität, analytische Leistung und klinische Leistung – gelten natürlich auch für CDx.
- Die wissenschaftliche Validität sollte auch die klinische Performance des assoziierten Arzneimittels in der entsprechenden, durch das CDx stratifizierten/selektierten Patientenpopulation verdeutlichen.
- Die analytische Leistungsbewertung sollte vor der Initiierung des Clinical Trial des Arzneimittels abgeschlossen sein (vgl. Punkt 2b “Ablauf der Co-Entwicklung”).
- Für den Nachweis der klinischen Leistung sind klinische Leistungsstudien erforderlich.
Weitere Informationen zu diesem Thema hat MedTech Europe zusammengestellt.
Ansonsten gelten keine besonderen Anforderungen an die Leistungsbewertung von CDx. Dennoch wird die EMA im Rahmen des Konsultationsverfahrens bestimmten Informationen besondere Aufmerksamkeit widmen:
“The application dossier should contain sufficient information about the scientific validity and/or scientific rationale for the use of the biomarker, device measurement characteristics, device development characteristics, analytical and clinical performance.”
Quelle: Guidance on the procedural aspects for the consultation to the European Medicines Agency by a notified body on companion diagnostics
Informationen zur Leistungsbewertung haben wir Ihnen in einem gesonderten Beitrag zusammengestellt.
Labelling
Die Gebrauchsanweisung des CDx muss den internationalen Freinamen (INN) des dazugehörigen Arzneimittels, für das es sich um einen therapiebegleitenden Tests handelt, enthalten.
Im Beipackzettel des Arzneimittels wird das CDx gemäß deutscher Gesetzgebung nicht angegeben (AMG §11).
b) Besonderheiten in den USA
Die US-Behörden haben bereits mehr Erfahrung mit CDx als die europäischen Behörden bzw. Benannte Stellen. In den USA bestehen es die folgenden regulatorischen Besonderheiten:
- Das gesamte Verfahren wird über die FDA abgewickelt und nicht wie in Europa über zwei Behörden (Benannte Stelle und EMA).
- Die meisten CDx sind “High-risk Devices” und werden der Klasse III zugeordnet, was mit der Auflage zur Einreichung einer Premarket Application (PMA) verbunden ist.
Nur wenige CDx können über die 510(k) Application zugelassen werden.
- Die FDA erlaubt im Kontext onkologischer Fragestellungen, dass CDx auch für eine spezifische Gruppe von Arzneimitteln vorgesehen sein können und nicht nur für einzelne Arzneimittel. Voraussetzung ist, dass hinreichend Evidenz vorliegt, dass das IVD für diese Gruppe von Arzneimitteln geeignet ist.
Zu diesem Thema gibt es das Guidance-Dokument Developing and Labeling In vitro Companion Diagnostic Devices for a Specific Group of Oncology Therapeutic Products der FDA.
- In den USA gibt es eine List of Cleared and Approved Companion Diagnostics.
- Labelling: Die Verwendung von CDx zusammen mit einem therapeutischen Produkt muss in den USA in der Gebrauchsanweisung sowie der Kennzeichnung des Diagnostikums UND des entsprechenden therapeutischen Produkts erwähnt werden (vgl. die Ausführungen der FDA hierzu).
Wie die FDA Sie bei der Klassifizierung unterstützt, erfahren Sie in unserem Fachartikel.
c) Leitlinien (oder der Mangel daran)
Derzeit mangelt es noch an Leitlinien zum Thema CDx. Die folgenden empfehlenswerten Dokumente liegen aber schon vor:
- Guidance on the procedural aspects for the consultation to the European Medicines Agency by a notified body on companion diagnostics der EMA
- Clinical Evidence Requirements for CE certification under the In-Vitro Diagnostic Regulation in the European Union der MedTech Europe
- MDCG 2020-16 rev. 2 Guidance on Classification Rules for in vitro Diagnostic Medical Devices under Regulation (EU) 2017/746 (insbesondere Annex II)
- MDCG 2022-10 Q&A on the interface between Regulation (EU) 536/2014 on clinical trials for medicinal products for human use (CTR) and Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR)
- Concept Paper der EMA
- Die Webseite der EMA liefert zudem Infos zur Implementierung sowie weitere Informationen zur Rolle der EMA, inklusive Antragsformular und Berichtsvorlage. Hierzu gab es aber keine Update seit Februar 2019.
- Die ausstehende sechste Revision der Guideline on the clinical evaluation of anticancer medicinal products der EMA soll eine bessere Leitlinie zu den regulatorischen Erwartungen für Biomarker-gesteuerte Entwicklung liefern
- FDA-Guidance zu onkologischer Forschung und CDx
- FDA-Empfehlung zu Co-Devo: Die Entwicklung von Biomarker-basierter diagnostischer Methoden sollte frühzeitig in der klinischen Entwicklung des Arzneimittels bedacht werden. Das Ziel ist die Maximierung der klinischen Anwendbarkeit der Technologie.
- MedTech Europe und die European Federation of Pharmaceutical Industries and Associations (EFPIA) haben ein Dokument verfasst, das die regulatorischen Unklarheiten von CDx auflistet und Lösungsvorschläge aufführt
Aus Mangel an konkreten europäischen Vorgaben empfehlen wir, sich vorerst an den Vorgaben der FDA zu orientieren. Zum Thema “Clinical Evidence” sind die Infos von MedTech Europe hilfreich. Außerdem sollten sich Hersteller immer über die aktuellen Entwicklungen informieren. Hierzu können sie beispielsweise den Regulatory Radar des Johner Instituts nutzen.
4. CDx auf den europäischen Markt bringen
Damit Sie ihr therapiebegleitendes Diagnostikum sicher auf den Markt bringen, können Sie sich an den üblichen Schritten der Zulassung orientieren. Beachten Sie dabei jedoch jeweils die Besonderheiten von CDx.
a) Schritt 1: Zweckbestimmung als CDx festlegen
Prüfen Sie anhand der Zweckbestimmung Ihres In-vitro-Diagnostikums, ob es sich überhaupt um ein CDx handelt. Dies ist nur der Fall, wenn tatsächlich eine Abhängigkeit zum therapeutischen Mittel besteht (vgl. Punkt 1, “Definition”).
b) Schritt 2: Qualitätsmanagementsystem
Im nächsten Schritt setzen Sie Ihr Qualitätsmanagementsystem nach ISO 13485 und IVDR auf. Hierbei gibt es KEINE Unterschiede zu anderen Typen von IVD.
c) Schritt 3: Technische Dokumentation zusammenstellen
Auch bei der Zusammenstellung der Technischen Dokumentation nach den Anhängen II und III der IVDR gibt es keine Besonderheiten im Vergleich mit anderen IVD. Die Anforderungen der II und III der IVDR sind zu erfüllen. Beachten Sie unsere Hinweise zur Leistungsbewertung (vgl. Punkt “Regulatorische Besonderheiten”).
Während der Produktentwicklung sollten IVD-Hersteller in Austausch mit dem pharmazeutischen Partner stehen.
d) Schritt 4: Konformitätsbewertung
Das Konformitätsbewertungsverfahren ist aufwendiger als bei anderen IVD (vgl. Punkt “Konsultation der Arzneimittelbehörde”). Es startet mit der formellen Antragstellung bei der Benannten Stelle. Hier müssen Sie neben dem Namen und der Klassifizierung des CDx u. a. auch die spezielle Zweckbestimmung inklusive dem Verweis auf das betreffende Arzneimittel nennen.
Die Arzneimittelbehörde, in der Regel die EMA, wird durch die Benannte Stelle einbezogen.
Die Benannte Stelle kann in Austausch mit der Arzneimittelbehörde treten. Wenn erforderlich, kann ein Pre-Submission Meeting einberufen werden, das die Vorbereitung des Antrags unterstützt. Hieran nehmen EMA (Rapporteur), Benannte Stelle und ggf. der Marktzulassungsinhaber/Antragsteller des Arzneimittels teil. Daran anschließend erfolgt die Übermittlung des Antrags durch die Benannte Stelle und das Konsultationsverfahren beginnt. Es folgt dem oben beschriebenen Ablauf (Abschnitt 3 a).
5. Tipps für Entwicklung und Zulassung von CDx
Damit bei der Entwicklung und Zulassung Ihres therapiebegleitenden Diagnostikums alles glatt läuft, haben wir folgende Empfehlungen für Sie:
a) Partner gut aussuchen
Seien Sie bei der Wahl Ihres Partners aus dem Bereich IVD oder Pharma ruhig kritisch. Die Entwicklung eines CDx erfordert ein enormes Maß an Abstimmung und Zusammenarbeit. Dazu sollten die Rahmenbedingungen stimmen. Leichter wird das CDx-Projekt, wenn Ihr Partner bereits Erfahrung mit therapiebegleitenden Diagnostika hat.
b) Klare Absprachen treffen
Funktionierende Absprachen und ein hohes Level an Kommunikation sind wichtig, um die Kompatibilität von IVD und Therapeutikum zu gewährleisten.
Die Planung sollte dahingehend abgestimmt sein, dass die IVD-Entwicklung mit den Anforderungen hinsichtlich des Arzneimittels übereinstimmt.
Die Prototypentwicklung inklusive der analytischen Leistungsbewertung sollte so koordiniert sein, dass rechtzeitig bzw. planmäßig das Clinical Trial gestartet werden kann.
Die EMA empfiehlt hierzu:
“The Agency recommends early interactions with the relevant notified body, the device manufacturer, and the marketing authorisation holder(s) or applicant(s) of the medicinal product(s) (as applicable and relevant).”
Quelle: Guidance on the procedural aspects for the consultation to the European Medicines Agency by a notified body on companion diagnostics
Die FDA empfiehlt hierzu:
“Although codevelopment as a process does not require simultaneous development of the IVD companion diagnostic and the therapeutic product from beginning to end, the availability of an IVD with “market-ready” analytical performance characteristics (i.e., a test that is completely specified with complete analytical validation and meets the therapeutic product sponsor’s expectations for performance) is highly recommended at the time of initiation of clinical trial(s) intended to support approval of the therapeutic product.”
Quelle: https://www.fda.gov/media/99030/download
c) Beziehen Sie das CDx früh in die Arzneimittelentwicklung ein
Die Abstimmung von Therapeutikum und CDx sollte so früh wie möglich beginnen. Daher ist es wichtig, dass Arzneimittelhersteller den IVD-Hersteller so früh wie möglich einbezieht.
d) Planen Sie mehr Zeit als üblich ein
Wenn ein therapiebegleitendes Diagnostikum ein Arzneimittel begleitet, sind sowohl die Entwicklung als auch die Zulassung aufwändiger als bei einem Arzneimittel oder einem IVD allein. Auch die Beteiligung der Arzneimittelbehörde kostet Zeit. Diese Faktoren sollten Sie unbedingt in Ihrer Planung berücksichtigen.
6. Fazit
Therapiebegleitende Diagnostika (CDx) werden in der modernen Medizin immer wichtiger. Insbesondere in der personalisierten Medizin spielen sie eine große Rolle.
Für Hersteller von IVD und Arzneimitteln bedeuten sie jedoch mehr Aufwand; CDx sind mit zusätzlichen regulatorischen Anforderungen verbunden. Entscheidend ist daher eine gute Planung und eine effektive Abstimmung zwischen allen Beteiligten. Wenn dies gelingt, kann das CDx einen enormen Nutzen bringen und die Therapie für Patientinnen und Patienten auf ein neues Level heben.
Die Expertinnen und Experten des Johner Instituts unterstützen Sie gerne bei Fragen zur Zulassungsstrategie von IVD oder speziell zum Thema therapiebegleitende Diagnostika (CDx). Kontaktieren Sie uns einfach über das Formular oder schreiben Sie eine E-Mail.
Versionshistorie:
- 15.12.2023: Kapitel 3 c) hinzufügen von MDCG 2020-16 rev. 2 und MDCG 2022-10
- 28.2.2023: Kapitel 1 b) Weiterführende Informationen ergänzt um Verlinkung auf MDCG 2020-16 rev.2
- 7.1.2022: Guidance on the procedural aspects for the consultation to the European Medicines Agency by a notified body on companion diagnostics der EMA ergänzt.


Guten Tag Frau Dr. Bartsch,
Sie schreiben unter 3 a) „Alle Companion Diagnostics fallen in die Risikoklasse C.“ Das entspricht IVDR Anhang VIII Regel 3. Werden in der IVDR Artikel 48(3) Absatz 3 und 48(4) Absatz 2 nicht auch therapiebegleitende Diagnostika der Klasse D adressiert? Oder verstehe ich den Text an der Stelle falsch?
Vielen Dank und beste Grüße
Sehr geehrter Herr Graß,
Sie liegen richtig, mit Klasse C beziehe ich mich auf Regel 3(f) des Anhangs VIII der IVDR.
Die Aufführungen von IVDs in Artikel 48 (3) und (4) entsprechen Aufzählungen besonderer Anforderungen während der Konformitätsbewertung. Diese beziehen sich auf Klasse D-Produkte Produkten zur Eigenanwendung, für patientennahe Tests und therapiebegleitende Diagnostika.
Sollte ein IVD mehrere Zweckbestimmungen haben, von denen eine einem CDx-Produkt und eine weitere einem Klasse-D-Produkt entspricht, würde Implementierungsregel 1.8. zum Tragen kommen. Demgemäß würde das gesamte Produkt aufgrund der mehrfachen Zweckbestimmung in die höhere Klasse D fallen.
Die Klassifizierung von IVDs erfolgt gemäß Anhang VIII der IVDR. Gemäß Implementierungsregel 1.7. sind alle Implementierungs- und Klassifizierungsregeln des betreffenden Anhangs zu beachten, um die richtige Klassifizierung des IVDs vorzunehmen.
Für Klasse D-Produkte kommen Regel 1 und 2 in Frage, wobei sich Regel 2 auf IVDs zur Blutgruppenbestimmung und Untersuchungen im Zusammenhang mit Transfusions- und Transplantationsmedizin bezieht, „um die Immunkompatibilität von für die Transfusion, Transplantation oder Zellgabe bestimmtem Blut, Blutbestandteilen, Zellen, Geweben oder Organen festzustellen“. Regel 1 Gedankenstrich 1 und 2 beziehen sich auf Produkte deren Zweck der Nachweis übertragbarer Erreger ist, um zum einen „ihre Eignung für die Transfusion, Transplantation oder Zellgabe zu bewerten“ und zum anderen „die eine lebensbedrohende Krankheit mit einem hohen oder mutmaßlich hohen Verbreitungsrisiko verursachen“.
Der dritte Gedankenstrich von Regel 1 verweist auf den Zweck zur „Bestimmung der Infektionslast einer lebensbedrohlichen Krankheit, dessen Überwachung im Rahmen des Patientenmanagements von Bedeutung ist“.
Definitionsgemäß stimmen diese Zweckbestimmungen nicht mit dem Zweck eines therapiebegleitenden CDx gemäß IVDR überein (s. Abschnitt 1 a), graue Box des Artikels).
Allerdings sollten wir dazu noch einmal Regel 1, Gedankenstrich 3 besonders betrachten. Man könnte in diesem Zusammenhang beispielsweise an Antibiotikabehandlungen denken. Aber der benannte Zweck bezieht sich klar auf die „Überwachung“. Die IVDR legt jedoch in Erwägungsgrund (12) explizit fest, dass „Produkte, die zur Überwachung der Behandlung mit einem Arzneimittel eingesetzt werden“ nicht als CDx gelten.
Die MedTech Europe geht ebenfalls davon aus, dass voraussichtlich alle CDx in die Klasse C fallen: „However, as all CDx devices are expected to be in class ‘C’, […]“ (Clinical Evidence Requirements for CE certification under the In-Vitro Diagnostic Regulation in the European Union der MedTech Europe).
Vielen Dank für Ihre Geduld.
Beste Grüße
Sophie Bartsch