
Ein Design Change ist eine Änderung der Auslegung eines Produkts. Es ist wichtig zu verstehen, welche regulatorischen Implikationen daraus entstehen und wo es unter Umständen einen Einfluss auf die Gültigkeit der Konformitätserklärung des Produkts gibt.
Dieser Artikel gibt einen Überblick und löst damit viele derzeit gängige Missverständnisse auf.
Damit Sie schnell und sicher entscheiden können, ob ein Design Change im Sinne der Übergangsregeln der EU-Verordnungen (MDR/IVDR) signifikant ist, und Ärger mit Behörden vermeiden können.
1. Design Change: Worum es geht
1.1 Definition
Die deutsche Ausgabe der MDR/IVDR übersetzt „Design Change“ als „Änderungen der Auslegung“ des Produkts. Immer dann, wenn sich am Entwurf des Medizinprodukts etwas ändern würde, läge eine Design-Änderung vor.
Unter Design Change versteht man somit nicht (nur) die Änderung des (grafischen) Designs eines Produkts.
Vielmehr versteht man darunter jede Änderung des Entwurfs eines Produkts vor oder nach dessen jeweiliger Freigabe.
Beachten Sie, dass MDR und IVDR auch eine Änderung der Zweckbestimmung als Design Change auslegen, auch wenn das Produkt unverändert bleibt.
1.2 Beispiele
Von einer Design-Änderung/einem Design Change spricht man beispielsweise, wenn der Hersteller das Folgende ändert:
- das Layout einer Platine
- die Benutzer-Produkt-Schnittstelle (das „User Interface“)
- die Funktionen, die das Produkt anbietet (weil dadurch etwas an der Auslegung geändert werden muss)
- Leistungsangaben z. B. die Nachweisgrenze eines Immunoassays oder die Energieabgabe eines HF-Chirurgiegeräts
- Materialien, aus denen das Produkt gefertigt ist
Wenn ein Software-Entwickler feststellt, dass die bereits freigegebene Architektur einen Fehler enthält und diese ändert (implizit im Code oder/und explizit im Architektur-Dokument), dann liegt auch ein Design Change vor.
Wie bereits erwähnt, zählen MDR und IVDR auch eine geänderte Zweckbestimmung, z. B. neue Indikationen oder eine andere Anwendergruppe, als Design Change.
2. Regulatorische Anforderungen
2.1 Anforderungen der FDA an Design Changes
Die FDA hat festgestellt, dass man bei Problemlösungen oft neue Probleme einführt. Deshalb fordert sie, dass die Hersteller diese „Lösungen“ (Änderungen) sehr sorgfältig bewerten, bevor sie sie realisieren.
Konkret schreibt der 21 CFR § 820.30(i) „Design changes“:
Each manufacturer shall establish and maintain procedures for the identification, documentation, validation or where appropriate verification, review, and approval of design changes before their implementation.
21 CFR § 820.30(i)
In der Diskussion dieses Paragrafen weist die FDA auf Folgendes hin:
- Stellen Sie sicher, dass die Dokumente entsprechend überarbeitet und gelenkt (z. B. versioniert und freigegeben) werden.
- Stellen Sie durch Verifizierung und Validierung sicher, dass das Problem, das mit diesem Design Change gelöst werden soll, auch wirklich gelöst ist.
- Achten Sie darauf, dass die Änderungen genehmigt sind und bewertet wird, dass keine neuen Probleme durch die Änderung verursacht wurden und die bisherigen Anforderungen weiterhin erfüllt werden.
- Kommunizieren Sie die Änderungen, damit andere Entwicklungsabteilungen, die Produktion, der Vertrieb und die Kunden Bescheid wissen.
Lesen Sie hier mehr zum Thema Guidance Document ‚Deciding When to Submit a 510(k) for a Change to an Existing Device‘.
2.2 ISO 13485 zu Design- und Entwicklungsänderungen
Auch in Europa gibt es Anforderungen an den Umgang mit „Design Changes“. So fordert die ISO 13485 in Kapitel 7.3, die Design- und Entwicklungsänderungen zu lenken.
Die Forderungen gleichen denen der FDA: Die Änderungen müssen
- genehmigt,
- bewertet,
- verifiziert und validiert und
- dokumentiert werden.
2.3 MDR/IVDR
Sowohl die EU-Medizinprodukte-Verordnung MDR als auch die EU-Verordnung über In-vitro-Diagnostika (IVDR) gehen an mehreren Stellen auf Design-Änderungen ein, beispielsweise in:
- MDR Artikel 10 (9) / IVDR Artikel 10 (8): „Änderungen an der Auslegung […] werden zeitgerecht angemessen berücksichtigt.“
- MDR Anhang VI, Teil C, 6.5.2 / IVDR Anhang VI, Teil C, 6.2.2 (Software): „Eine neue UDI-DI ist immer dann erforderlich, wenn Folgendes geändert wird: die ursprüngliche Leistung; die Sicherheit oder die bestimmungsgemäße Verwendung der Software; die Auswertung der Daten. Zu diesen Änderungen gehören neue oder geänderte Algorithmen, Datenbankstrukturen, Betriebsplattformen und Architekturen oder neue Schnittstellen oder neue Kanäle für die Interoperabilität.“
- MDR Anhang IX, 4.10 / IVDR Anhang IX, 4.11: „Änderungen an dem genehmigten Produkt müssen von der Benannten Stelle, die die EU-Bescheinigung über die Bewertung der technischen Dokumentation ausgestellt hat, genehmigt werden, wenn diese Änderungen die Sicherheit und Leistungsfähigkeit des Produkts oder die für das Produkt vorgeschriebenen Anwendungsbedingungen beeinträchtigen könnten.“
3. Wesentliche Design Changes: Wann eine Änderung meldepflichtig gegenüber der Benannten Stelle ist
Jeglicher geplante Design Change muss gemäß ISO 13485, Kapitel 7.3 innerhalb des QM-Systems bewertet werden. Im Folgenden beschreiben wir, wann zusätzlich die Benannte Stelle einbezogen werden muss.
3.1 Guidance Document des TEAM NB
Die Vereinigung der Benannten Stellen (Team NB) hat mit der NB-MED/2.5.2/Rec2 eine Empfehlung publiziert, die etwas mehr Klarheit zur Kommunikation von Design Changes an die jeweilige Benannte Stelle schaffen soll.
Darin beschreiben die Autoren, wann eine Design-Änderung als wesentlich („substantial/significant“) zu bewerten und damit meldepflichtig gegenüber der Benannten Stelle ist. Das wäre jede Änderung am Produkt, die die Konformität mit den grundlegenden Anforderungen oder den vom Hersteller festgelegten Anwendungsbereich bzw. Kontraindikationen beeinflussen könnte.
Konkret benennt das Dokument Änderungen
- der Zweckbestimmung, Indikation, Kontraindikation;
- von Leistungsmerkmalen;
- eines Zulieferers(!);
- aufgrund von Risiken, die noch nicht betrachtet wurden;
- von Warnungen;
- der vorgesehenen Nutzergruppen;
- der vorgesehenen Nutzung;
- von Charakteristiken, die durch die klinische Bewertung noch nicht berücksichtigt sind;
- als Folge von Überlegungen, die aus der Marktbeobachtung stammen, inklusive aus Zwischenfällen, Rückrufen oder Beschwerden;
- getrieben durch State-of-the-Art Entwicklung (z. B. neuste Technologien);
- die die Produktion beeinflussen;
- die die Sicherheit und Leistungsfähigkeit des Produkts betreffen.
Das Ziel der Meldepflicht dabei ist es, der Benannten Stelle die Möglichkeit zu geben, die Konformität des Produkts nach einer Änderung des Designs zu prüfen. Benannte Stellen können aufgrund einer gemeldeten Produktänderung entscheiden, ob sie eine erneute Ad-hoc-Prüfung der Konformität durchführen oder die Änderung im nächsten planmäßigen Audit betrachten.
Das TeamNB-Dokument stellt an dieser Stelle nur eine allgemeingültige Interpretation von substantiellen/signifikanten Änderungen dar. Da das Dokument noch unter der MDD/IVDD erstellt wurde, stellt es unter der MDR/IVDR lediglich eine Hilfestellung dar. Rechtlich bindend ist ohnehin nur der Vertrag zwischen dem Medizinproduktehersteller und seiner Benannten Stelle, in dem die Meldekriterien und -bedingungen explizit und individuell definiert sind.
3.2 Scope des Zertifikats
Entscheidend ist zudem der Anwendungsbereich des Zertifikats: Falls Sie Ihr Produkt, das Sie über Anhang II der MDD bzw. über Anhang IX der MDR in den Verkehr bringen, so ändern, dass es nicht mehr in den Scope fällt, dürfen Sie das nicht, ohne die Benannte Stelle einzubeziehen.
Die Zertifikate erlauben Ihnen als Hersteller gerade, Produkte innerhalb des Zertifikats zu entwickeln und in den Verkehr zu bringen, ohne die Benannte Stelle um Erlaubnis zu fragen. Allerdings verlangen die Benannten Stellen häufig, informiert zu werden.
4. Signifikante Design Changes (Artikel 120 MDR / 110 IVDR, MDCG 2020-3)
4.1 MDR / IVDR
Von den bisher beschriebenen Meldepflichten für Design Changes gegenüber den Benannten Stellen muss ein weiteres Konzept komplett losgelöst betrachtet werden.
Signifikante Änderungen im Zusammenhang mit der Übergangsfrist von den europäischen Richtlinien (MDD/IVDD) zu den Verordnungen (MDR/IVDR) unterliegen anderen regulatorischen Implikationen. Hier geht es um die Frage, ob die bisherige Konformitätserklärung bei einer Änderung am Produkt gültig bleibt.
Im Kontext der Übergangsregelungen spielen die Design-Änderungen in den Verordnungen an folgenden Stellen eine Rolle:
MDR
By way of derogation from Article 5 of this Regulation, a device with a certificate that was issued in accordance with Directive 90/385/EEC or Directive 93/42/EEC and which is valid by virtue of paragraph 2 of this Article may only be placed on the market or put into service provided that from the date of application of this Regulation it continues to comply with either of those Directives, and provided there are no significant changes in the design and intended purpose.
MDR, Artikel 120(3)
IVDR
By way of derogation from Article 5 of this Regulation, the devices referred to in the second and third subparagraphs of this paragraph may be placed on the market or put into service until the dates set out in those subparagraphs, provided that, from the date of application of this Regulation, those devices continue to comply with Directive 98/79/EC, and provided that there are no significant changes in the design and intended purpose of those devices.
IVDR, Artikel 110(3)
Als Hilfestellung zur Bewertung der Änderungen am Produkt hat die MDCG zwei Leitlinien veröffentlicht. Hierin werden Kriterien genannt, unter denen die Änderungen als signifikant zu sehen ist und die Konformitätserklärung somit ihre Gültigkeit verliert.
4.2 MDCG 2020-3
Im März 2020 hat die MDCG die „MDCG 2020-3“ mit dem Titel „Guidance on significant changes regarding the transitional provision under Article 120 of the MDR with regard to devices covered by certificates according to MDD or AIMDD“ veröffentlicht. Diese Ausgabe wurde später überarbeitet, dabei um hilfreiche Beispiele ergänzt und im Mai 2023 als MDCG 2020-3 Rev.1 herausgegeben.
4.2.1 Nicht-signifikante Änderungen
Die MDCG stellt fest, was sie nicht als signifikante Design Changes versteht:
- Administrative Änderungen
- Name und Adresse des Herstellers
- Rechtsform, z. B. von GmbH nach GmbH & Co. KG
- Bevollmächtigter
- Organisatorische Änderungen
- Neue Fertigungsstätten, Umzug von Fertigungsstätten
- Neue oder geänderte Lieferanten und Dienstleister
- Im gewissen Umfang auch Änderungen am QM-System
- Einschränkung der Zweckbestimmung
- Beschränkung von Indikationen, klinischen Anwendungen oder der Patientenpopulation
- Korrekturmaßnahmen
- Nicht-signifikante Änderungen am Produkt, die sich nicht nachteilig auf die Leistung des Produkts auswirken, darunter Änderungen, die produktinterne Kontrollmechanismen, das Funktionsprinzip, die Energieversorgung oder ggf. Alarmsysteme nicht ändern
Für nicht-signifikante Änderungen, die explizit das Nutzen-/Risikoverhältnis nicht negativ beeinflussen, gibt die Leitlinie folgende Beispiele:
Spezifikation/Kennzeichnung
- Änderungen, die den zertifizierten Bereich von Spezifikationen enger fassen, wie die Schraubenlänge innerhalb der bisherigen Länge oder Durchmesser
- Änderung des Griffs eines steuerbaren Ablationskatheters, um den ergonomischen Komfort für das medizinische Personal oder das ästhetische Erscheinungsbild eines Geräts zu verbessern
- Umformatierung einer bestehenden Bedienungsanleitung
- Änderung des Label- oder des Verpackungsmaterials eines nicht-sterilen Produkts
Komponenten
- Austausch eines Halbleiterbauelements/einer elektronischen Komponente/einer elektronischen Baugruppe mit denselben Spezifikationen
- Aufbringen einer Beschichtung auf eine berührungslose Leiterplatte zur Verbesserung der Stromisolierung
- Änderung der Größe oder der geometrischen Form der Bedienungs-/Alarmtasten
Energieversorgung
- Wechsel eines Batterietyps
- Wechsel der Batteriechemie
- Wechsel des Ladegeräts oder des Netzkabels bei gleicher Spezifikation
Materialien
- Neuer oder zusätzlicher Lieferant/Hersteller eines Materials im Rahmen der festgelegten Spezifikationen
- Substitution eines chemischen Stoffes, um anderen geltenden Gesetzen und Verordnungen zu entsprechen, z. B. der REACH-Verordnung
- Ersetzen eines Stoffes oder Materials, die das Nutzen-Risiko-Verhältnis nicht negativ beeinflussen, wie
- Verbesserung der Materialqualität ohne Änderung der Spezifikationen
- Ersetzen eines Materials der Verpackung eines sterilen Produkts, das nicht zur Aufrechterhaltung der Sterilbarriere beiträgt
- Änderung eines Materials, das lediglich als Verarbeitungshilfsmittel und somit als Herstellungs- und nicht als Konstruktionsänderung gilt
Sterilität
- Änderung der Parameter des Sterilisationszyklus, unter der Voraussetzung, dass es keine Auswirkungen auf das Sterilitätssicherungsniveau oder die Sterilisationsrückstände gibt
- Verlängerung der Haltbarkeitsdauer, validiert durch von der benannten Stelle genehmigte Verfahren
- Umstellung von einfach steriler auf doppelt sterile Verpackung
Laut MDCG 2020-3 können sich Hersteller von den Benannten Stellen bestätigen lassen, dass eine Designänderung nicht signifikant ist. Das stellt aber kein ergänzendes Zertifikat dar.
4.2.2 Signifikante Änderungen
Die MDCG definiert folgende Änderungen als „signifikant“:
- Erweiterung und Änderungen der Zweckbestimmung, z. B. neue Indikationen, neue Patientenpopulation, andere klinische Anwendung
- UI-Änderungen, die weitere Usability-Daten bedürfen
- Änderungen der Leistungsspezifikation
- Änderung der Produktabmessungen oder Konstruktionsmerkmale außerhalb der aktuellen Spezifikationen, wie neue Stentlängen, die außerhalb des Bereichs der zuvor zertifizierten Stentlängen liegen oder Hinzufügen von Elektroden an einem implantierbaren Herzschrittmacherkabel
- Ausweitung der Spezifikationsgrenzen für kritische Komponenten oder Parameter
- Einsatz neuer Sensoren mit unterschiedlichem Funktionsprinzip, wie ein Luftblasendetektor mit Ultraschall im Vergleich zu einem optischen Lichtsensor
- Designänderungen, die entweder Risiken erhöhen oder bestehende risikominimierende Maßnahmen betreffen
- Entfernen oder Hinzufügen eines Alarmsystems oder Handhabung einer Alarmsituation
- Umstellung von analoger auf digitale Steuerung
- Wechsel von einem manuellen zu einem softwaregesteuerten Gerät
- Austausch oder Änderungen von Materialien, es sei denn, sie stammen von bestehenden Lieferanten und finden innerhalb unveränderter Spezifikationen statt.
- Wechsel von einem Material mit einem geringen toxikologischen oder biologischen Risiko zu einem Material mit einem höheren Risiko
- Hinzufügung oder Veränderung eines Materials menschlichen/tierischen Ursprungs
- Änderungen am Sterilisationsprozess oder an der Verpackung, die einen Einfluss auf die Sterilität haben können
- Änderung der abschließenden Sterilisationsmethode
- Wechsel eines Produkts von der Kennzeichnung „unsteril“ zur Kennzeichnung „steril“
- Verlängerung der Haltbarkeitsdauer, die nicht anhand von durch die benannte Stelle genehmigten Protokollen validiert ist
- Viele Software-Änderungen
Lesen Sie hier mehr zum Thema Design Changes bei Software und zu Beispielen für Software-Änderungen.
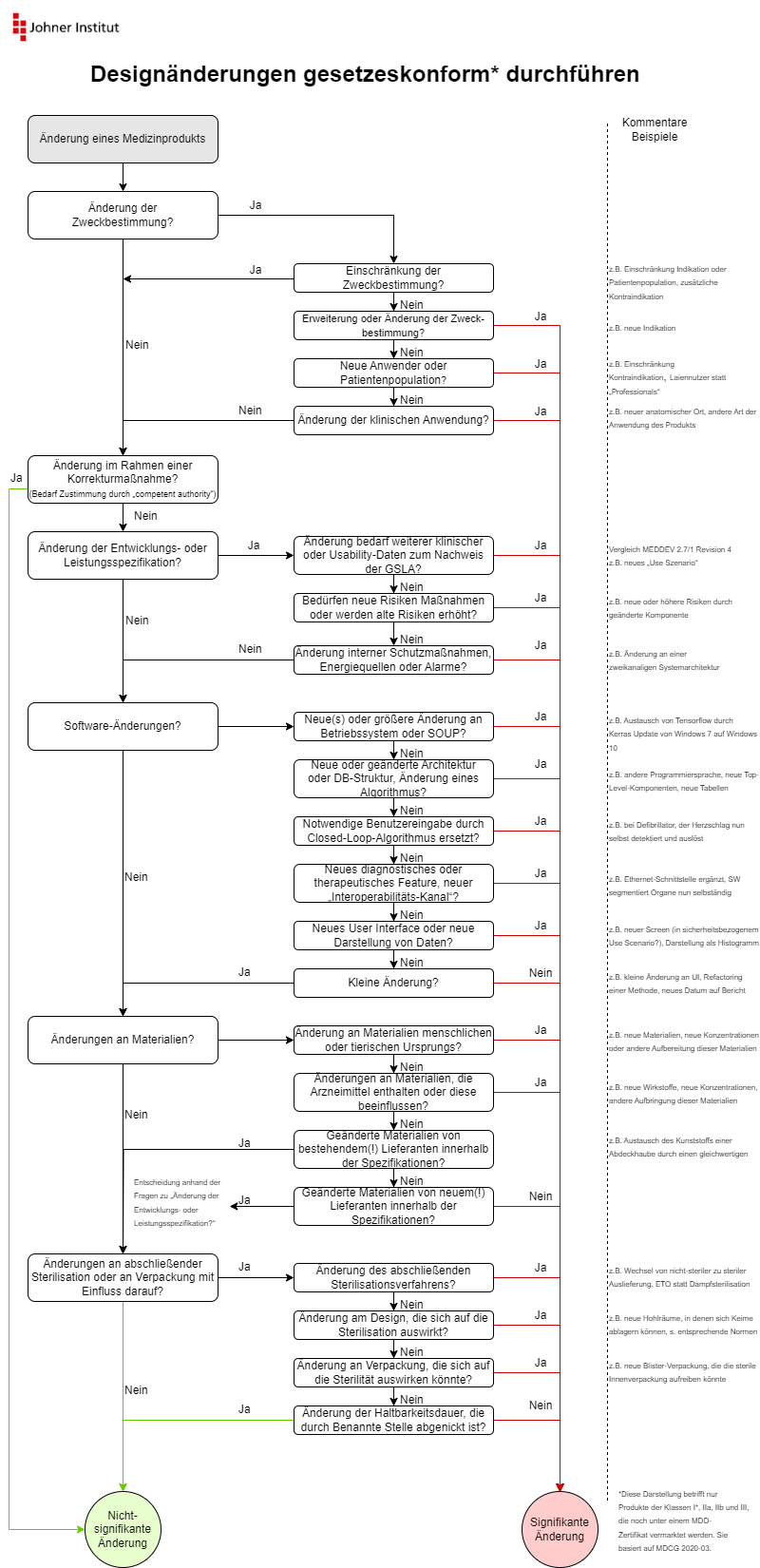
Die MDCG beschreibt die Änderungen in Form von Flussdiagrammen. Die Infografik des Johner Instituts fasst diese Diagramme auf einer Seite zusammen und ergänzt sie um Beispiele.
Sie können diese Infografik kostenlos herunterladen:
4.2.3 Kritik
Das MDCG-Dokument ist hilfreich. Durch die Überarbeitung wurden formale Mängel behoben und außerdem viele Beispiele ergänzt, welche den inhaltlichen Nutzen der Leitlinie verbessert haben.
Einige Fragen lässt das jedoch offen. Besonders zu den folgenden Fragen hätte man sich eine Antwort gewünscht:
- Will man wirklich alle Korrekturmaßnahmen (wohlgemerkt Korrekturmaßnahmen, nicht Korrekturen) auch dann durchwinken, wenn damit nennenswerte Änderungen am Design einhergehen? Die Reihenfolge des Flussdiagramms lässt genau das vermuten.
- Weshalb will man Korrekturmaßnahmen durchwinken, Korrekturen aber nicht? Bei der Software deklariert man Bugfixes nicht als „signifikant“.
- Soll jede Änderung an einer database structure als signifikant eingestuft werden? Jede neue Spalte in einer Datenbank?
4.3 MDCG 2022-6
Nach der Leitlinie MDCG 2020-3 für Medizinprodukte unter MDR wurde für In-vitro-Diagnostika im Mai 2022 die Leitlinie MDCG 2020-6 unter dem Titel „Guidance on significant changes regarding the transitional provision under Article 110(3) of the IVDR“ von der MDCG herausgegeben. Sie konkretisiert, welche Änderungen eines Produkts, das den Übergangsfristen der IVDR unterliegt, als signifikant einzuordnen sind und baut dabei auf den Festlegungen der Leitlinie MDCG 2020-3 auf.
Somit gelten die generellen Angaben sowohl für nicht-signifikante Änderungen, wie administrative oder organisatorische Änderungen, als auch für signifikante Änderungen, wie Erweiterungen der Zweckbestimmung oder Änderungen in der Leistungsspezifikation, auch für die Betrachtung von IVD. Darüber hinaus grenzt die MDCG in der Leitlinie MDCG 2022-6 jedoch konkreter und anhand von Beispielen signifikante von nicht-signifikanten Änderungen nach Art der Änderung voneinander ab:
4.3.1 Änderung der Zweckbestimmung
Nicht-signifikant
- Einschränkung der Zweckbestimmung, wie durch Beschränkung der Patientengruppe
Signifikant
- Jegliche Erweiterung der Zweckbestimmung
- Änderung der Zweckbestimmung, wie eine Änderung des Assay-Typs von einem qualitativen zu einem quantitativen Assay oder einer Änderung des Probentyps
4.3.2 Designänderungen
Nicht-signifikant
- Designänderungen, die das Funktionsprinzip des Produkts nicht verändern, also die Testmethode oder das Nachweisverfahren; darunter fällt beispielsweise eine Veränderung eines Probenaufreinigungsschritts oder der Austausch eines PCR-Cyclers
Signifikant
- Designänderungen, bei denen das Funktionsprinzip des Produkts verändert werden, wie der Wechsel der Testmethode von Immunfluoreszenz zu ELISA oder von einer fotometrischen zu einer chromatografischen Messung
- Jegliche Änderung, die sich nachteilig auf die Leistung des Produkts auswirkt. Das heißt: Alle beschriebenen nicht-signifikanten Änderungen müssen darauf hin überprüft werden, ob sie sich nachteilig auf die Leistung auswirken, und werden dadurch ggf. zu einer signifikanten Änderung.
4.3.3 Änderungen, die Software betreffen
Nicht-signifikant
- Bugfixes (auch hier muss die Auswirkung auf Sicherheit und Leistung überprüft werden)
- Updates bestehender Laufzeitumgebungen, wie Windows
- Verbesserungen der Benutzerschnittstelle
Signifikant
- Änderungen in der Laufzeitumgebung, wie einem Wechsel des Betriebssystems
- Modifizierungen der Software-Architektur oder des Algorithmus
4.3.4 Materialänderungen sowie Modifikationen des Sterilisationsverfahrens
Nicht-signifikant
- Änderungen in Bezug auf Materialien, die nicht maßgeblich für das Funktionsprinzip sind, wie Ersetzen eines Konservierungsmittels oder Austausch von Chemikalien zur Konformität mit der REACH-Verordnung (falls keine Beeinträchtigung der Leistung besteht)
- Abänderung der Parameter des Sterilisationszyklus im Rahmen eines zertifizierten QMS
Signifikant
- Änderungen in Bezug auf Inhaltsstoffe und Materialien, die für das Funktionsprinzip wesentlich sind, wie PCR Primer, Antikörper oder Antigene bei Immunoassays oder Marker bei der Chromatografie
- Änderung der Sterilisationsmethode, was insbesondere beinhaltet, ein nicht-steriles Produkt in ein steriles abzuändern
- Verpackungsänderungen, die sich negativ auf die Gewährleistung der Sterilität auswirken können
4.4 Sonderfall Software
Dieses Kapitel fasst die gesetzlichen Anforderungen und Leitlinien für Software zusammen und geht im Wesentlichen mit den Beispielen von MDCG 2020-3 und MDCG 2022-6 einher.
4.4.1 Signifikante Software-Änderungen
Eine Design-Änderung wäre bei Software wahrscheinlich dann signifikant, wenn eine der folgenden Bedingungen vorliegt:
- Es gibt eine Änderung an der Zweckbestimmung, einschließlich
- vorgesehener Nutzungsumgebung,
- vorgesehenen Nutzergruppen,
- neuen oder anderen Indikationen,
- weniger Kontraindikationen (also eine Erweiterung der Zweckbestimmung).
- Sie beseitigen einen Fehler in der Software oder der zugehörigen Gebrauchsanweisung, um Risiken zu minimieren (> Rückruf).
- Sie ändern die Benutzer-Produkt-Schnittstelle (unwesentliche Änderungen wie Korrektur von Rechtschreibfehlern ausgenommen). Das trifft insbesondere dann zu, wenn Sie
- Warnmeldungen hinzufügen, ändern oder entfernen,
- neue sicherheitsbezogene Use Scenarios einfügen,
- kritische UI-Elemente (ehemals Hauptbedienfunktionen) ändern.
- Sie verwenden eine neue Technologie, z. B. ein neues Framework, neue SOUPs (gemeint ist nicht eine neue Version), oder gar eine andere Programmiersprache.
- Die Software soll in einer anderen Laufzeitumgebung (Betriebssystem oder Version, Prozessor, Bildschirmgröße-/auflösung) eingesetzt werden.
- Die Software muss eine neue Datenschnittstelle bedienen.
- Die Entwickler fügen in der Datenbank neue Tabellen oder Fremdbeziehungen ein.
- Die Entwickler ändern einen zentralen Algorithmus oder ersetzen Nutzereingaben durch einen Algorithmus, der als Regelkreis dient, z. B. zur Berechnung von Medikamentendosen, zur Bestrahlungsplanung oder für die Bildbearbeitung.
- Das betrifft insbesondere den Austausch von konventionellen Algorithmen durch KI-Algorithmen bzw. den Austausch eines KI-Modells (z. B. neuronales Netzwerk durch ein Boosting-Verfahren).
4.4.2 Nicht-signifikante Software-Änderungen
Üblicherweise zählt man die folgenden Änderungen an einer Software nicht als meldepflichtig:
- Security-Patches und Updates bestehender Laufzeitumgebungen, wie Windows Updates
- Update einer SOUP durch neuere Version
- Kleine Refactorings, z. B. innerhalb einer Methode
- Hinzufügen eines Attributs zu einer Datenbank oder Änderung des Datentyps eines Attributs
- Anpassung der Software, um mit neuen Versionen einer bestehenden Schnittstellenspezifikation arbeiten zu können (z. B. Update von HL7 V2.7 auf 2.8)
- Kleine Maßnahmen zur Verbesserung nicht-funktionaler Eigenschaften wie der Robustheit (z. B. zusätzliche Werteüberprüfung)
5. Fazit und Zusammenfassung
5.1 Es ist verwirrend, was mit Design Changes gemeint ist
Für viele Hersteller ist es sehr verwirrend, dass es im Grunde drei Betrachtungsebenen bei Produktänderungen gibt, die in diesem Artikel beschrieben werden:
- Design Changes als solches, mit den entsprechenden Prozess- und Dokumentationspflichten
- Die Entscheidung, welche der Änderungen an die Benannte Stelle kommuniziert werden müssen
- Für den Fall, dass noch eine gültige Konformitätserklärung gegen die Richtlinien MDD/IVDD besteht: Die Entscheidung, ab wann die Konformitätserklärung durch eine Produktänderung erlischt
Unglücklicherweise verwendet man für die beiden letzten Änderungskriterien gleichzeitig die Bezeichnung “significant change” bzw. “substantial change”.
Die Unterscheidung der MDR und IVDR zwischen Software-Änderungen, die eine Änderung der UDI-DI bedingen, und jenen, die „nur“ eine Änderung der UDI-PI zur Folge haben, mag bei der Unterscheidung zwischen meldepflichtigen und nicht-meldepflichtigen Änderungen dienlich sein: Eine Änderung der UDI-PI ist üblicherweise nicht meldepflichtig.
5.2 Es gibt Unterstützung
Das Johner Institut empfiehlt im Zweifelsfall, mit der Benannten Stelle eine klare Absprache zu treffen.
Die MDCG bietet mit ihren Dokumenten 2020-3 und 2022-6 eine gute Hilfestellung, um zwischen signifikanten und nicht-signifikanten Design Changes im Zusammenhang mit der Gültigkeit der Konformitätserklärungen beim Übergang zu den Verordnungen zu unterscheiden.
Mit der überarbeiteten 2020-3 für Medizinprodukte unter MDR und mit der 2022-6 für IVD sind ausgereifte Leitlinien vorhanden, die konkrete Beispiele an Änderungen beinhaltet.
Im eingeschränkten Umfang steht Herstellern, Behörden und Benannten Stellen das kostenlose Micro-Consulting für solche Einschätzungen zur Verfügung.
Änderungshistorie
- 2025-02-25: Überschriftenebenen korrigiert, Bedeutung der MDCG 2020-3 im Text gestärkt. Text mit Zwischenüberschriften besser gegliedert, Überschriften neu nummeriert
- 2023-08-15: Vollständige Überarbeitung des Artikels
- 2022-06-23: Im letzten Hinweis auch die Produkte der Klasse I* ergänzt.
- 2022-01-02: Poster um Hinweis ergänzt, dass es sich „nur“ auf Produkte der Klasse IIa und höher bezieht, die unter einem MDD-Zertifikat in den Verkehr gebracht werden.
- 2021-07-19: In Poster letzte Entscheidung korrigiert (enthielt eine Verneinung zu viel)
- 2021-06-06: In Poster Entscheidung bei „Materialien von neuem Lieferanten innerhalb der Spezifikation“ korrigiert (Danke an O. Tan für Hinweis!)
- 2021-03-27: In Poster Hinweis auf „Competent Authority“ ergänzt
- 2021-01-20: Unterkapitel zu TEAM-NB Dokument zu „drug device combination products“ ergänzt
- 2020-01-26: Hinweis direkt über Kapitel 5 ergänzt



{kind=link}
Guten Tag,
Vielen Dank für Ihren Artikel, allerdings stellt sich mir eine Frage: Welche Änderungen sind dann kein Design Change wenn auch z.b. die Farbe der Verpackung eine Designänderung wäre?
Vielen Dank für Ihre Antwort.
MFG
Dr. Breß
Sehr geehrter Herr Dr. Breß,
wie groß eine Änderung sein darf, damit Sie nicht als Design Change gilt, hängt vom Risikomanagement und der Art der Änderung ab. Beispielsweise gibt Ihnen die MDR bei SW einen Hinweis: Die Änderungen, die nur einer neuen Product Identification bedürfen, aber keiner neuen Design Identification, könnte man als ausreichend klein charakterisieren.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
zu Ihrer Antwort vom Dienstag 17. Juli 2018 um 17:19 bräuchte ich eine Aufklärung: Was meinen Sie mit der „Design Identification“? In der MDR kommt dieser Begriff nicht vor. Nur „Device Identifyer“. Eine neue UDI-DI braucht man schon, wenn sich die Version der SW ändert. Und die ändert sich bereits bei kleinster Änderung.
Wo befindet sich dieser Hinweis für SW in der MDR?
Für eine Rückmeldung wäre ich sehr dankbar.
Mit freundlichen Grüßen
Natalie Gudd
Sehr geehrte Frau Gudd,
da scheine ich mich vertippt zu haben. Ich meinte natürlich mit DI den Device Identifier.
Die Hinweise auf sie Software finden Sie im Anhang VI, Teil C 6.5.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
Vielen Dank für die ausführliche Zusammenfassung!
Leider ist Ihnen beim Erstellen des Design-Change Posters ein Fehler unterlaufen. Die Beschriftung der Pfeile abgehend von Software-Änderungen?->Kleine Änderung? ist genau andersrum. Kleine Änderungen sind, wie im Artikel richtig beschrieben, nicht signifikant.
Mit freundlichen Grüßen,
Helmut Steiner
Sie haben absolut Recht, lieber Herr Steiner!
Die neue Version lade ich gleich hoch.
Besten Dank!
Herzliche Grüße, Christian Johner
Guten Tag,
gilt die Änderung des Produktnamens nach 26.05.2021 als signifikant? Dadurch ergeben sich einige nötige Anpassungen, allerdings bleibt das unter MDD zertifizierte Produkt ansonsten identisch.
Vielen Dank,
L. Borchert
Sehr geehrte Frau Borchert,
die MDCG 2020-03 ist hier ein relevantes Dokument. Sie erfüllen keine der dort genannte Kriterien. Damit ist der Change als nicht signifikant einzuordnen.
Allerdings wäre dieser in der EUDAMED zu hinterlegen.
Ich würde mir die Änderung von Ihrer Benannten Stelle schriftlich bestätigen lassen, um späteren Ärger zu vermeiden, da auf dem Zertifikat ein anderer Produktname steht.
Viele Grüße, Christian Johner
Guten Morgen Herr Johner,
In Ihrer Infografik Design Change v6 ist dargestellt, das alle Designänderungen im Rahmen einer Korrekturmaßnahme als nicht siginifkant zu bewerten sind. Laut MDCG-2020-03 müssen diese Korrekturmaßnahmen aber von der zuständigen Competent Authority akzeptiert worden sein. Zu einer solchen Bewertung durch die Competent Authority kommt es meiner Meinung nach nur bei einer Korrekurmaßnahme im Feld auf Grundlage eines Vorkomnisses.
Mit freundlichen Grüßen
Daniel Grabner
Sehr geehrter Herr Grabner, danke für Ihren Hinweis!
Ihr Hinweis betraf die Anmerkung im MDCG-Dokument zu den „competent authorities“. Diesen habe ich im Poster ergänzt.
Nochmals vielen Dank!
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
herzlichen Dank für die gute Zusammenfassung!
Ich habe eine Frage bzgl. dem Bewertungsbaum in der MDCG 2020-03, speziell zu den Materialänderungen.
Die Frage D4 „Ingredient ormaterial from new suppliermeetsexisting specification“ (Originaltext !) verzweigt bei Beantwortung mit ja zurück in die Bewertung B bei Vorliegen eines „Design or performance change“.
Mir erschließt sich der Sinn dieser Rückverweisung nicht, denn falls ich einen Change hätte, auf den Chart B zutrifft, wäre ich doch beim Abarbeiten des Main Charts eh schon in diesen eingetreten? Welchen Sinn hat dann diese Doppelbewertung?
Oder habe ich da ein grundlegendes Verständnisproblem?
Herzlichen Dank & freundliche Grüße
Olaf Kessel-Deynet
Sehr geehrter Herr Kessel-Deynet,
eine ausgezeichnete Frage! Was genau in den Köpfen der Autoren vor sich ging, weiß ich auch nicht. Daher kann ich nur spekulieren.
Möglicherweise wollten die Autoren, dass man für den neuen Supplier nochmals überlegt, ob es Risiken gibt, die speziell für diesen Supplier untersucht oder/und beherrscht werden müssen. Beispielsweise hat der Supplier ein anderes Fertigungsverfahren oder ein anderes QS-System. Dann müsste man die Risikoüberlegung nochmals anstellen.
Ihr Gedanke, dass man sich diese Überlegungen auch beim ersten Durchlauf bereits hätte machen könnte, ist dennoch logisch.
Viele Grüße, Christian Johner
Guten Tag,
danke für die ausführliche Erklärung. Mir ist dabei eine Frage aufgekommen, wir möchten eine neue Primärverpackung für unsere Augentropfen implementieren, so neu ist die Primärverpackung auch nicht, da wir sie bereits bei anderen Produkten einsetzen. Wir würden dies als nicht signifikante Änderung einordnen, was halten Sie davon?
Es gibt keinerlei Änderungen in Performance oder Sicherheit geschweige den Risiko, wir haben sogar das selbe Produkt mit gleicher Primärverpackung, nur selbstverständlich anderer Zweckbestimmung. Bisher ist es beim NB nicht durchgekommen.
Danke und Viele Grüße
V. Frank
Sehr geehrte Frau Frank,
danke für Ihre Frage. Ich fürchte, dass sich diese mit den gegebenen Informationen allein nicht beantworten lässt. Beispielsweise weiß ich nicht, ob die Sterilität bei diesen Augentropfen eine Rolle spielt (was ich vermute).
Eine Änderung einer Verpackung kann durchaus eine signifikanten Änderung darstellen. Wenn diese eine Auswirkung auf die Sterilisation haben kann, dann sagt das die MDCG im Dokument 2020-3 auf Seite 14 sogar explizit. Aber auch andere Änderungen, die eine Auswirkung z.B. auf die Gebrauchstauglichkeit haben, sind u.U. signifikant.
Die Tatsache, dass eine Verpackung wo anders bereits verwendet wurde, ist für die Antwort nicht erheblich. Sie ist aber dienlich, dass die grundlegenden Sicherheits- und Leistungsanforderungen auch nach der Änderung erfüllt sind, unabhängig davon, ob diese signifikant sind.
Viele Grüße, Christian Johner
Hallo Herr Prof. Johner,
vielen Dank für die super ausführliche Zusammenfassung 🙂
Meine Frage an Sie ist, ob Sie in Bezug auf ein Softwareprodukt (App) einen Wechsel des Cloud-Providers (Backend) als einen signifkanten Desing Change einstufen, auch wenn die SW-Architektur und Infrastruktur-Architektur gleich bleibt und es keine Quellcode-Änderungen (außer dem Austausch der URL) gibt?
Vielen Dank vorab für Ihre Einschätzung und viele Grüße
T. Becker
Sehr geehrter Herr Becker,
danke für die spannende Frage!
Die Antwort hängt davon ab, was Sie in der Zweckbestimmung bzw. den Markt- und Software-Anforderungen formuliert haben. Wenn Sie dort sagen, dass es auf einem beliebigen Cloud-Dienst laufen kann, wäre es überhaupt kein Change. Denn der Cloud-Dienst ist idealerweis als Laufzeitumgebung definiert und nicht als Teil des Produkts.
Ein Austausch des „vorderen Teils“ der URL sehe ich ebenso nicht als wesentliche Änderung. Beim hinteren Teil (Routing) würde ich es anders sehen. Denn das ist defacto die externe Schnittstelle des Produkts.
Herzliche Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
ich habe eine spannende und wichtige Frage an Sie zum Thema signifikante Änderung beim MPe-Herstellerwechsel, bei dem die Meinung und das „Bauchgefühl“ vieler Verbände und anderer Sachkundiger gespalten ist – mit der mehrheitlichen Tendenz, es sei eine signifikante Änderung, was ich aber überhaupt nicht verstehe. Vor allem, weil außer dem „Bauchgefühl“ keine Belegstelle genannt wird, das MDCG Flowchart wird nicht als Referenz genannt. Aber wo kommt die Auffassung dann her?
Es geht konkret um den folgenden Fall:
Ein Lohnhersteller stellt ein Medizinprodukt (Klasse 1 MDD, derzeit legacy device) für einen juristischen Hersteller her, auf den dieses Medizinprodukt derzeit registriert ist.
Nun würde der juristische Hersteller aber seine Position als Inhaber gerne an den Lohnhersteller übertragen – weil ihm die „Luft“ ausgegangen ist.
Diese Änderung würde, bis auf die Änderung des Herstellers auf dem Packmittel (Industriehüttensymbol), eine Erstanzeige im DMIDS und eine „neue Konformitätserklärung“ des Lohnherstellers betreffen, aber das Medizinprodukt selbst in keiner Weise verändern.
In Abschnitt 3b) bzw. auch in der MDCG 2020-03 wird eine solche Änderung als administrative und nicht meldepflichtige Änderung bewertet. Jedoch ist die MDCG nicht rechtlich bindend.
• Woher aber kommt dann die oben beschriebene Auffassung, es sei dennoch eine signifikante Änderung?
• Kann ein Hersteller eine solche Änderung selbst als nicht signifikant festlegen und den Sachverhalt vertraglich festhalten?
• Kennen Sie ggf. ähnliche Präzedenzfälle?
• Könnte ein solcher administrativer Hersteller-Wechsel die Übergangsfrist des Medizinprodukts mit Verkaufsmöglichkeit bis 27.05.2025 gefährden?
• Stammt das „Bauchgefühl“ bzw. die Auffassung der Verbände aus anderen Übertragungsfällen, bei denen es sich nicht um Klasse 1 Medizinprodukte gehandelt hat und es wäre daraus als mitgeltende Info eine Feststellung darüber zu erhalten, dass es sich um eine signifikante Änderung handelt, sofern der Fall tatsächlich dann auch auf Klasse 1 MPe übertragbar ist.
Vielen herzlichen Dank im Voraus!
Mit freundlichen Grüßen
I. Milke
Lieber Herr (oder Frau) Milke,
der Hintergrund des Themas ist folgender: Die Benannten Stellen müssen fortlaufend Produkte und Hersteller überwachen. Sie tun dies in der Regel einmal jährlich über Audits. Wenn der Hersteller zwischen den Audits etwas an seinem System oder seinen Produkten ändert, musste er schon unter der MDD die Benannten Stelle über eine solche „signifikante Änderung“ unterrichten, damit diese entscheiden konnte, ob sie zwischenzeitlich nochmals eine Überprüfung macht oder die Änderung erst im nächsten Routine-Audit überprüft. Nun hat die Einführung der MDR die Benannten Stellen gezwungen, ihre eigenen Betriebsprozesse komplett auf MDR umzustellen. Aus diesem Grund hat der Gesetzgeber über die MDR festgelegt, dass in dieser Übergangszeit beim Hersteller keine Änderungen mehr durchgeführt werden dürfen, die ein erneutes Überprüfen durch eine Benannte Stelle erforderlich macht.
Wenn Sie diese Überlegungen als Basis nehmen, fällt es Ihnen vielleicht leichter, den Sachverhalt zu bewerten. Stellen Sie sich vor, die Produktverantwortung geht tatsächlich an eine andere legale oder juristische Person über. Funktioniert dann das Meldewesen noch (Vigilanz)? Wird vom neuen Verantwortlichen die Marktüberwachung weiterhin korrekt durchgeführt? Kann er entsprechende Prozesse vorweisen und sind die konform? Gibt es damit verbundene Änderungen in den Prozessen Beschaffung, Service, Lagerung und Transport?
Genau das müsste in diesem Fall durch eine Benannte Stelle geprüft und bewertet werden, was man aber aus den genannten Gründen über den Artikel 120 verhindern will. Wobei eine signifikante Änderung ja nicht verboten ist, sie würde dann nur unweigerlich zu einer Neu-Bewertung des Produktes unter der MDR führen.
Eine alleinige Änderung des Namens eines Herstellers ist übrigens berechtigterweise eine „administrative“ Änderung. Die Änderung des Namens, weil der verantwortliche Hersteller wechselt, ist jedoch nicht mehr als „administrativ“ zu bezeichnen.
Noch eine Anmerkung: Ich nehme an, Sie sprechen in Ihrem Beispiel von Klasse I*-Produkten (steril oder mit Messfunktion), denn die anderen Produkte der Klasse I (ohne Benannte Stelle) fallen gar nicht erst unter die Übergangsbestimmungen des Artikel 120. Sie mussten sofort seit Mai 2021 konform mit der MDR sein und die Überlegung zu „signifikanten Änderungen“ gelten demnach für diese Produkte gar nicht.
Herzliche Grüße
Christian Rosenzweig
Sehr geehrter Herr Prof. Johner,
im Guidance Dokument MDCG 2022-6 steht, dass die Änderung der Sterilisationsmethode eine signifikante Änderung ist. Wie würden Sie in diesem Fall „Sterilsationsmethode“ definieren? Ist es eine signifikante Änderung wenn man von gamma auf x-Ray Sterilisation ändert? In diesem Fall würde für mich die Sterilisationsmethode gleich bleiben, da es sich in beiden Fällen um Sterilisation durch Bestrahlung handelt. Der Sterilisationsdienstleister ist ebenfalls in beiden Fällen der gleiche.
Eine signifikante Änderung wäre für mich der Wechsel von Gamma auf ETO Sterilisation.
Ich bin gespannt auf Ihre Einschätzung!
Besten Dank im Voraus!
Schöne Grüße,
Martina
Liebe Martina,
der Zweck der MDCG 2022-6 ist es, zu entscheiden, wann man mit der Benannten Stelle über eine „Verletzung“ des Zertifikates und damit über die Neuzertifizierung des Produktes unter der IVDR sprechen muss. Ich würde argumentieren, dass die Umstellung eines Sterilisationsverfahrens (Gamma auf X-Ray) die Frage aufwirft, ob das Produkt mit den neuen Bestrahlungsparametern und Bedingungen noch immer genauso sicher ist. Immerhin wird vermutlich eine komplett andere Infrastruktur im Bestrahlungsprozess eingebunden und eine neue Prozessvalidierung dafür aufgesetzt. Um die Bestätigung der Konformität des Produktes durch die Benannte Stelle kommen Sie in diesem Fall also nicht herum und damit bewegen Sie sich bereits im Bereich der Konformitätsbewertung unter der IVDR.
Zwei Tipps an dieser Stelle:
a) Sprechen Sie das mit Ihrer Benannten Stelle ab
b) Planen Sie auch ohne diese Überlegung so früh wie möglich Ihren Umstieg auf die IVDR, denn bald sind die Kapazitäten der Benannten Stellen mehr als ausgeschöpft!
Herzliche Grüße
Christian Rosenzweig
Sehr geehrter Herr Prof. Johner,
wieso wurde ihr FlowChart (Poster-Design-Change-V13) dahingehend angepasst, dass er sich jetzt nur noch auf Produkte der Klasse IIa und höher bezieht? Produkte der Klasse I*, für welche ein MDD-Zertifikat durch die benannte Stelle ausgestellt wurde sind doch ebenfalls vom Artikel 120 der MDR betroffen und müssen entsprechend bewertet bzw. Änderungen gemeldet werden.
Mit freundlichen Grüßen
Stefan
Sehr geehrter Stefan, danke für Ihren Hinweis!
Sie haben Recht. Es geht um alle Produkte, die eine Benannte Stelle benötigen nach MDR. Und da zählen die Klasse I* Produkte dazu.
Ich verbessere das. Nochmals vielen Dank!
Viele Grüße
Guten Tag,
wir haben eine Frage zu dem Kapitel 5.2 Quality system changes des „Guidance for manufacturers and Notified Bodies on reporting of Design Changes and Changes of the Quality System“.
Kann man hier sagen, dass Änderungen am Qualitätsmanagementsystem immer mit einer Design Änderung zusammenhängen?
Viele Grüße
Burgert
Liebe/r Frau/Herr Burgert,
bei Änderungen am Qualitätsmanagementsystem möchte die Benannte Stelle informiert werden, um entscheiden zu können, ob die Bescheinigung aufrechterhalten bleiben kann, bzw. ob sie sich mit einem erneuten Audit von der Konformität überzeugen muss. Da das QM-System aber fortlaufend angepasst und verbessert werden muss (Anforderung der MDR bzw. der ISO 13485), wären fortlaufend Benachrichtigungen an die Benannte Stelle notwendig.
Generell gilt deshalb die Aussage in diesem Guidance-Dokument auf der ersten Seite unten:
Wenn Sie beispielsweise im QM-System Ihren Entwicklungsprozess verändern, dann kann es sein, dass Sie auch den Prozess zur Produktänderung mit anfassen und dann könnte das später einen wesentlichen Einfluss auf das bereits freigegebene Produkt-Design haben. Dann wäre diese QMS-Änderung also benachrichtigungspflichtig gegenüber der Benannten Stelle. Am Design des Produktes selbst haben Sie zu diesem Zeitpunkt nichts verändert. Also hängen Änderungen am QMS nicht notwendigerweise mit Produktänderungen zusammen und sind völlig getrennt zu betrachten.
Beantwortet das Ihre Frage?
Herzliche Grüße
Christian Rosenzweig
Guten Tag,
in meiner Firma kommt es häufig zu Diskussionen bezüglich Design Change und Obsoleszenz von Materialien und Bauteilen.
Wie auch Ihnen bekannt ist, muss auf Grund der aktuellen Lage immer öfters alternativen für Bauteile und Materien gefunden werden. Wir dokumentieren dies über unser Änderungsmanagement und kommen immer wieder zu Diskussionen, ob es eine Designänderung ist oder nicht. Ein Beispiel wäre der Austausch eines elektrischen Bauteils auf einer Leiterplatte mit gleichen Spezifikationen (sprich die Leiterplatte muss nicht angepasst werden). Kann mit der Argumentation, dass die Spezifikationen und / oder die Funktionalität gleichbleiben gesagt werden, dass es sich um Obsoleszenzhandling handelt?
Und die wichtigere Frage: Wo besteht die Grenze zwischen normaler Änderung und Design Change? Und kann man Design Changes als geringer signifikant ein ordnen um den Auffand geringer zuhalten?
Viele Grüße
P. Schmitt
Liebe Frau Schmitt,
zunächst muss jede Änderung sorgfältig über einen Änderungsprozess laufen, um überhaupt in der Lage zu sein, Entscheidungen über die nötigen Aktivitäten (inklusive Meldepflichten) treffen zu können. Das betrifft das Design des Produktes, die Fertigung oder Bereiche wie die Beschaffung oder den Service. Damit hätten wir die grundsätzliche Dokumentationspflicht für Änderungen geklärt, die unabhängig vom Grund einer Änderung ist.
Als Nächstes können wir zur Meldepflicht einer Änderung festhalten, dass die unter Umständen vielfältige Aspekte hat. Bei einem Klasse III Implantat verlangt die Benannte Stelle von Ihnen vielleicht, dass JEDE Änderung dorthin gemeldet wird, also im Prinzip auch der spezifikationsgerechte Tausch eines Elektronikbausteins. Ich habe in der Praxis schon erlebt, dass Elektronikkomponenten angeblich spezifikationsgerecht getauscht wurden und später hat sich herausgestellt, dass zwar die technischen Betriebsparameter gleich waren, aber die Lebensdauer beim neuen Baustein viel geringer war. Oder die zur Verfügung gestellten neuen Elektronikkomponenten waren aus Schrott ausgelötet, weil es eben im Moment eine absolute Knappheit gibt, und damit war die Komponente unerkannt vorgeschädigt. Nachvollziehbar, dass die Benannten Stellen den Herstellern bei kritischen Produkten genau auf die Finger schauen möchten.
Es kann auch sein, dass der Lieferant als „kritisch“ eingestuft ist und die Benannte Stelle hat eine Liste Ihrer kritischen Lieferanten und braucht Information, wenn Sie einen Lieferanten wechseln. Auch das kann bei einer einfachen Obsoleszenz eine Rolle spielen.
Und dann bleibt noch die Frage nach einer „wesentlichen“ Änderung im Sinne des Artikels 120 der MDR, wo es um die Frage geht, welche Änderungen am Produkt zu einem MDD-Zertifikats-Verlust führen. Da würde man einen spezifikationsgerechten Austausch einer Komponente nicht als „wesentliche“ Änderung ansehen. Das beruht auf den Entscheidungsdiagrammen im MDCG 2020-3. Frage „B“ im Main Chart und Frage D3 im Chart D sind da relevant.
Zusammengefasst:
Alle Änderungen müssen Sie immer in Ihrem Change-Prozess dokumentieren! Meldepflicht an die Benannte Stelle besteht gemäß deren vertraglichen Vorgaben. Das MDD-Zertifikat würde seine Gültigkeit nicht durch einen spezifikationsgerechten Ersatz einer nicht mehr verfügbaren Komponente verlieren.
Hilft Ihnen diese Erklärung?
Herzliche Grüße
Christian Rosenzweig
Guten Tag,
ich habe ebenfalls eine Frage bzw. ein konkretes Beispiel zu einer „Materialänderung“ und einer daraus eventuell resultierenden Meldepflicht. Konkret geht es um ein Klasse IIb Mittelohrimplantat, welches bisher aus Titan Grade 2 gefertigt wird. Aufgrund aktueller Lieferprobleme, soll das Produkt zukünftig aus Titan Grade 4 hergestellt werden. Die unterschiedlichen Titan Grade unterscheiden sich in ihrer prozentualen chemischen Zusammensetzung (innerhalb der ASTM F67 fest definiert).
Eine Argumentation lautet, dass es keine Änderung des Materials gibt (da beides mal Titan innerhalb der gleichen Norm spezifiziert), eine andere Argumentation wäre, dass es durchaus ein anderes Material ist (ähnlich verschiedenen Edelstahlsorte, z.B. 1.4301, 1.4021 etc.) Sterilisation, Herstellung, Verpackung, klin. Performance, Spezifikationen etc. bleiben unangetastet. Lediglich eine Änderung des Zulieferers für das Titan könnte sich (je nach weltweiter Verfügbarkeit) ändern.
Die Frage, die sich uns nun stellt ist, ob dies eine meldepflichtige Änderung darstellt oder nicht.
Ich bedanke mich für Ihre Hilfe und freue mich auf Ihre Antwort.
Mit freundlichen Grüßen
TaiTzu
Lieber TaiTzu,
ohne die fachliche Expertise und genauen Sachverhalte können wir Ihnen auf diesem Weg kaum eine valide Aussage geben. Aber den Weg kann ich Ihnen gerne aufzeigen:
Dokumentieren Sie schriftlich eine Argumentation, warum diese Änderung nach Ihrer Einschätzung basierend auf MDCG 2020-3 keine signifikante Änderung ist. Da Sie aber gemäß jeweiligem Vertrag mit Ihrer Benannten Stelle Änderungen am Produkt an diese kommunizieren müssen, reichen Sie die Änderung zusammen mit Ihrer Argumentation ein und lassen sich bestätigen, dass die Benannte Stelle Ihrer Argumentation folgt. Immerhin steht im MDCG 2020-3 „In case of doubt whether a change is significant they should ask their notified body.“
Herzliche Grüße
Christian Rosenzweig
Guten Tag,
Vielen Dank für den informativen Artikel. Ich habe eine konkrete Frage zur Bereitstellung der Gebrauchsanweisung. Ist ein Wechsle von einer gedruckten GBA zu einem elektronischen System eine signifikante Änderung im Sinner des Artikel 120 oder einfach nur eine Umsetzung von regulatorischen Möglichkeiten?
Falls die Umstellung auf ein elektronisches System als signifikante Änderung bewertet werden würde, wäre diese Umstellung für Legacy Devices nicht möglich und das obwohl die MDD dies mit der Verordnung 207/2021 erlaubt.
Mit freundlichen Grüßen
Clara Steinsiek
Liebe Frau Steinsiek,
pauschal lässt sich das nicht beantworten. Wenden Sie am besten das Entscheidungsdiagramm des MDCG 2020-3 systematisch an. In Teil B stoßen Sie zum Beispiel auf Fragen wie „Does the change require further clinical or usability data to support safety and performance?“. Die Gebrauchsanweisung stellt einen wesentlichen Teil der Gebrauchstauglichkeitsbetrachtung dar und wird deshalb häufig zum Gegenstand von Usability-Prüfungen. Insofern könnte – sofern Sie es in Ihrem Fall nicht anders belegen – die Antwort auf diese Frage „ja“ lauten und damit eine signifikante Änderung vorliegen.
Auch die Frage „Do new risks require control measures or are existing risks negatively affected? könnte mit „ja“ beantwortet werden. Sie müssen ohnehin bei der Umstellung auf eine elektronische Gebrauchsanweisung eine umfassende Risikoanalyse erstellen (Forderung der (EU) 2021/2226 in Absatz (4)) und können mit dieser Analyse den Beleg für Ihre Antwort auf die Frage des Guidance-Dokumentes erbringen.
Die Entscheidung, ob eine signifikante Änderung vorliegt, treffen Sie also auf Basis der dokumentierten Analyse des Entscheidungsdiagramms. Und diese Analyse können Sie dann auch der Benannten Stelle vorlegen, wenn Sie die Änderung durchführen möchten, denn gemäß Ihres Vertrages mit der Benannten Stelle müssen Sie vermutlich so eine Änderung in jedem Fall der Benannten Stelle mitteilen.
Hilft Ihnen das weiter?
Herzliche Grüße
Christian Rosenzweig
Guten Tag,
ich habe einen konkrete Frage zum Wechsel des OEMs von IVD legacy devices. Wir wollen bei einem IVD Kit zu einem anderen, bereits bestehenden OEM wechseln. Die Funktion sowie das Funktionsprinzip bleiben gleich. Die critical ingredients bleiben auch die gleichen, aber von einem anderen Rohstofflieferanten. Der Wechsel des OEMs hat keinen negativen Einfluss auf die Performance bzw. die Sicherheit oder das Risiko des IVDs. Die Zweckbestimmung bleibt ebenso unverändert. Die MDCG 2022-6 erlaubt einen Wechsel eines Lieferanten von Komponenten, sofern die Spezifikation unverändert bleibt. Können wir dann auch den OEM eines ganzen IVD-Kits wechseln, sofern die Produktspezifikation ident ist oder ist das dann als signifikante Änderung einzustufen?
Vielen Dank und viele Grüße,
J. Gross
Liebe Frau Groß,
wenn ich Ihrer Darstellung folge, dann ändert sich außer der Qualitätssicherungsvereinbarung (neuer Lieferant) an Ihrer bestehenden Technischen Dokumentation bezüglich der Produktauslegung nichts. In diesem Fall würde tatsächlich als nicht-signifikante Änderung die IVDD-Konformität aufrechterhalten bleiben.
Falls das Produkt schon über eine Benannte Stelle konformitätsgeprüft wurde: In der Regel ist die Änderung nach den spezifischen vertraglichen Vorgaben der Benannten Stelle dort anzeigepflichtig. Bei der Gelegenheit können Sie sich also bestätigen lassen, dass das Zertifikat tatsächlich erhalten bleibt.
Sorgen Sie in jedem Fall im Rahmen Ihrer Change-Dokumentation für ausreichend Nachweise, dass das Produkt weiterhin sicher und leistungsfähig ist.
Herzliche Grüße
Christian Rosenzweig
Thema MDCG 2020-03:
Wie lange hat man Zeit eine „non-significant“ Änderung zu implementieren, wenn diese Änderung von der Benannten Stelle genehmigt wurde, z.B. Namensänderung?
Haben Sie eine Idee zu den Fristen für die Implementierung?
Vielen Dank
Gruß
Sarah
Liebe Frau Hamed,
bei einer Konformitätsbewertung nach Anhang IX der MDR prüft die Benannte Stelle Ihr Qualitätsmanagementsystem. Man geht davon aus, dass alles, was Sie innerhalb dieses QMS durchführen, ordnungsgemäß umgesetzt wird. Deshalb sind Sie erstmal für alle Änderungen am Produkt selbst verantwortlich und selbst entscheidungsbefugt. Die Forderung, solche Änderungen an die Benannte Stelle zu melden, soll diesen die Möglichkeit geben, die Änderungen bei Bedarf von außen zu überwachen und über die Fortführung des bestehenden Zertifikats zu entscheiden. Die inhaltliche Umsetzung bleibt aber dennoch in Ihrer Hand und Verantwortung.
Das heißt also, wenn die Benannte Stelle Ihnen bereits signalisiert hat, dass Sie die Änderung durchführen können, dann ist es jetzt Ihre Entscheidung, wie schnell sie das tun. Natürlich würden Sie diese Änderungen bei sachlogischen Zwängen (z.B. Sicherheit gefährdet) oder bei geänderten regulatorischen Anforderungen (z.B. neue Grenzwerte oder Prozessanforderungen) zeitnah und dem Risiko angemessen umsetzen.
Eine Namensänderung Ihres Unternehmens sollte sich auch auf dem Label des Produktes oder in den Begleitpapieren zeitnah widerspiegeln. Genaue Fristen sind dafür aber nicht definiert.
Herzliche Grüße
Christian Rosenzweig
Guten Tag,
Ich hätte bezüglich einer Substitution eines Materials aufgrund von der REACH-Verordnung eine Frage: Und zwar, ist es notwendig bei solch einer Substitution die Biokompatibilitätstests neu durchzuführen, um die Konformität der Produkte zu gewährleisten? Muss man hier die Tests neu durchführen, um zu beweisen, dass die Änderung nicht substantiell sind? (Die Leistungsdaten bleiben gleich. Die biologische Sicherheit ist hier das Hauptaugenmerk)
Vielen Dank im Voraus!
Beste Grüße
Nam
Lieber Herr Nam Nguyen,
vielen Dank für Ihre Frage. Sie sehen das schon absolut richtig. So bald Sie ein Material des Medizinprodukts mit Kontakt zum Patienten (direkt oder indirekt) ändern, muss die Biokompatibilitätsbewertung aktualisiert werden. In den meisten Fällen müssen Sie in diesem Zusammenhang die Prüfungen neu durchführen lassen.
Herzliche Grüße
Sarah Gruber
Sehr geehrte Frau Gruber,
Vielen Dank für Ihre Antwort. Könnten Sie mir einen Fall erläutern, der keine erneute Biokompatibilitätsprüfung mit sich ziehen würde?
Herzliche Grüße
Nam Nguyen
Lieber Herr Nam Nguyen,
ein eindeutiger Fall wäre, wenn sich ein Material ohne Patienten ändern würde und die Änderung des Materials keinen Einfluss auf den Bereich des Medizinprodukts mit Patientenkontakt hat.
Ansonsten muss jeder Fall individuell betrachtet werden.
Wenn Sie gezielte Unterstützung benötigen, melden Sie sich gerne hinsichtlich eines Beratungskontingents bei uns.
Herzliche Grüße
Sarah Gruber
Hallo liebes Johner-Team,
wir vertreiben IVDs der Klasse A (steril), die wir derzeit noch unter der IVDD in Verkehr bringen. Ein Kunde möchte eines unserer Produkte als private gelabeltes Produkt. D.h. das Produkt bleibt gleich, aber das Labelling (inkl. IFU) wird mit einem anderen Frimenlogo und anderen Markennamen sein.
Ist das Ihrer Meinung nach ein neues Produkt, für welches die IVDR-Übergangszeit nicht mehr gilt? Oder würden Sie das als nicht signifikante Änderung der Kennzeichnung bewerten?
Ich würde es als neues Produkt sehen, welches dann bereits unter die IVDR fällt. Ich bin mir jedoch nicht sicher, ob ich das zu streng sehe.
Würde mich über Ihre Sichtweise freuen.
Schöne Grüße,
Maximilian
Lieber Herr Fürhauser,
vielen Dank für Ihre interessante Frage bezüglich Ihres Produkts und den Übergangsbestimmungen zur IVDR.
Die von Ihnen beschriebene gewünschte Vorgehensweise ähnelt dem nach IVDD, Art. 9 (5) erlaubten PLM-OEM Konstrukt. Dieses ist jedoch nach IVDR nicht mehr möglich und würde die Herstellerpflichten für den PLM nach sich ziehen.
Ebenfalls lässt sich dieses Vorgehen, wie Sie erwähnen, unter dem Aspekt einer Produktänderung betrachten. Hierbei lässt sich das MDCG 2022-6 heranziehen, wonach gemäß Kapitel 4.2 „changes concerning the manufacturer’s organisation (administrative changes)“ wie „changes of the manufacturer’s name, address or legal form“ nicht als signifikante Änderungen angesehen werden. Allerdings gibt es hierbei keine Aussage über den Produktname, weshalb die Argumentation des Beispiels nicht direkt verwendbar ist.
Außerdem ist zu beachten, dass der Produktname (ggf. was Sie unter „Markenname“ verstehen) mit dem in der Konformitätserklärung (DoC) angegebenen Bezeichnung übereinstimmen muss. Die DoC darf nicht ohne Weiteres geändert werden, ohne deren Gültigkeit zu verletzen.
Um eine fundiertere Einschätzung geben zu können, wären also weitere Informationen zu Ihrem Produkt und der genauen Konstellation erforderlich. Sollten Sie eine tiefergehende Analyse wünschen, stehen wir Ihnen natürlich gerne für die Ausarbeitung einer konformen Vorgehensweise zur Verfügung.
Beste Grüße
Janos Hackenbeck
Hallo liebes Johner Team,
ich habe gleich zwei Fragen zu signifikanten Änderungen.
1. Ist die NBOG-Guidance 2014-3 auch auf MDR zugelassene Produkte anwendbar? Meines Wissens gibt es hier noch keine neue Guidance für MDR Produkte. Gibt es hierzu schon Pläne für eine neue Guidance?
2. Ein steriles Medizinprodukt hat vom NB eine MDR Zulassung, und für dieses Medizinprodukt ist nun eine Erhöhung der Shelf Life geplant. Bei der geplanten Erhöhung der Shelf Life werden dieselben Tests durchgeführt (Verpackung, Produkt etc.), wie bei der ursprünglichen Produktzulassung, nur eben mit erhöhten Alterungszeiten. Ist diese Änderung als signifikant zu betrachten? Ich beziehe mich hierbei auf die NBOG 2014-3 Guidance, die m.E. eine solche Änderung als nicht signifikant einstuft („A change in the shelf life is considered a substantial change, and an approval by the Notified Body is required, when the protocols and methods for determining shelf life have been changed or have not been reviewed in a previous approval.)
Vielen Dank für den Support im Voraus
Anke Blocher
Liebe Frau Blocher,
1. Aktuell gibt es noch keine direkte MDR/IVDR-spezifische Guidance, die die NBOG 2014-3 vollständig ersetzt. Bis zur Veröffentlichung entsprechender Dokumente lassen sich die bestehenden NBOG-Guidelines, die für MDD/IVDD gelten als Orientierungshilfe verwenden, um beispielsweise Rationalen abzuleiten. Hierbei müssen jedoch wiederum die spezifischen Anforderungen der MDR berücksichtigt werden.
Ähnlich lassen sich die MDCG Guidance Dokumente heranziehen, die sich allerdings spezifisch auf die Übergangsbestimmungen beziehen (MDCG 2020-3 bzw. MDCG 2022-6).
2. Sowohl NBOG 2014-3 als auch MDCG 2020-3, rev. 1 schätzen eine Verlängerung der „Shelf Life“, die gemäß Protokollen validiert wurde, welche von der Benannten Stelle genehmigt wurden, als nicht signifikant ein. Obwohl die Änderung demnach nicht signifikant wäre, ist es ratsam, die geplante Shelf Life-Erhöhung mit der Benannten Stelle zu besprechen. Die genannten Guidance Dokumente können hier als Argumentationsgrundlage verwendet werden und dabei darauf verwiesen werden, dass diese den aktuellen „Stand der Technik“ abbilden.
Beste Grüße
Janos Hackenbeck