
Die Verordnung (EU) 2025/327 über den europäischen Gesundheitsdatenraum (European Health Data Space, kurz EHDS) ist eine weitere europäische Verordnung, die Hersteller von Medizinprodukten und IVD betreffen kann.
Was diese Verordnung fordert und was diese Hersteller bis wann tun müssen, klärt dieser Artikel ebenso wie mögliche Vorteile des EHDS bzw. der EHDS-VO.
Dieser Artikel verwendet die Begriffe „Verordnung (EU) 2025/327“, „EHDS-Verordnung“ und „EHDS-VO“ synonym.
1. Ziele und Nutznießer des European Health Data Space (EHDS)
a. Übersicht
Vom European Health Data Space sollen verschiedene Parteien profitieren:
- Patienten: Die Patientinnen und Patienten sollen einen besseren Zugang und eine bessere Kontrolle über ihre Gesundheitsdaten bekommen und von einer besseren Gesundheitsversorgung profitieren. Dazu trägt auch ein grenzüberschreitender Datenaustausch bei.
- Gesundheitsdienstleister: Krankenhäuser, medizinisches Fachpersonal wie Ärztinnen und Ärzte sowie andere medizinische Dienstleister können auf vollständigere Daten zugreifen.
- Forschung: Auch die Forschung soll von diesen Daten profitieren und damit mehr Innovation und eine bessere Gesundheitsversorgung ermöglichen.
- Politik: Die politischen Entscheider verfügen über belastbare Daten, was schnelle und gezielte Entscheidungen erlaubt, beispielsweise im Falle einer Pandemie.
- Europäische Union: Der EHDS ist ein wesentliches Element bei der Schaffung einer starken und widerstandsfähigen europäischen Gesundheitsunion.
Auf ihrer Webseite zum EHDS verlinkt die EU weitere Informationsquellen, u. a. ein Factsheet, das die Vorteile des European Data Spaces benennt, auch die ökonomischen Vorteile.
b. Vorteile für Hersteller von Medizinprodukten und IVD
Ob die Hersteller den einheitlichen Rechts- und Technologie-Rahmen und die dort geforderte Interoperabilität als förderlich empfinden, ist derzeit noch unklar.
Allerdings ist zu erwarten, dass die Hersteller von den neuen rechtlichen Möglichkeiten profitieren, die Gesundheitsdaten zu nutzen („Sekundärnutzung“) für
- die „Sicherstellung hoher Qualitäts- und Sicherheitsstandards“ (Artikel 53 a)),
- die „wissenschaftliche Forschung […], die […] ein hohes Maß an Qualität und Sicherheit […] von Medizinprodukten sicherstellt“ (Artikel 53 e) sowie
- „das Trainieren, Testen und Bewerten von Algorithmen, auch in Medizinprodukten, In-vitro-Diagnostika, KI-Systemen und digitalen Gesundheitsanwendungen;“ (Artikel 53 e II)
Damit dürfte der EHDS zu einer wichtigen Quelle für die Post-Market Surveillance und den Post-Market Clinical Follow-up werden.
Der berechtigte Zugriff auf weitere Gesundheitsdaten ermöglicht es den Herstellern zudem, neue Geschäftsmodelle und Auswertemöglichkeiten zu entwickeln.
Medizinprodukte können das medizinische Fachpersonal besser mit Diagnose- oder Therapievorschlägen unterstützen, wenn diese auf einem umfangreicheren Datensatz basieren. Die EHDS-Verordnung stellt einen wichtigen Pfeiler für die personalisierte Medizin dar.
Medizinprodukte wie Medical Apps können sich eines umfangreichen Datenschatzes bedienen und Patientinnen und Patienten bei der Vorbeugung, Linderung und Heilung von Krankheiten unterstützen.
2. „Betroffene“ der EHDS-Verordnung
a. Übersicht
Die Verordnung (EU) 2025/327 legt verschiedenen Organisationen Pflichten auf. Dazu zählen:
- Hersteller von EHR-Systemen (Electronic Health Record Systems)
- Importeure, Bevollmächtigte und Händler dieser Systeme
- Hersteller von Medizinprodukten und IVD, falls deren Produkte Daten mit EHR-Systemen austauschen
- Hersteller von Wellness-Anwendungen, falls deren Produkte Daten mit EHR-Systemen austauschen
- Gesundheitsdienstleister und Angehörige der Gesundheitsberufe
- Europäische und nationale (Gesundheits-)Behörden
Diese Gruppen sind nicht überschneidungsfrei. Beispielsweise zählen einige EHR-Systeme auch als Medizinprodukte.
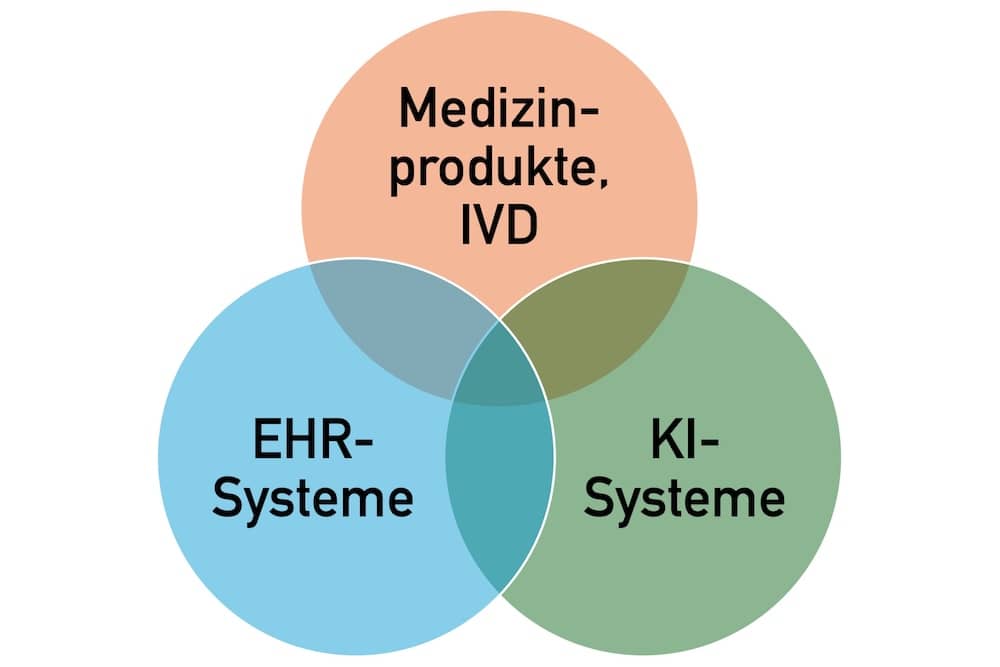
b. Hersteller von EHR-Systemen
Die Verordnung zum European Health Data Space definiert den Begriff „EHR-System“.
jedes System, bei dem die Software oder eine Kombination aus Hardware und Software des Systems es ermöglicht, personenbezogene elektronische Gesundheitsdaten, die zu den durch diese Verordnung eingeführten prioritären Kategorien personenbezogener elektronischer Gesundheitsdaten gehören, zu speichern, zu vermitteln, zu exportieren, zu importieren, zu konvertieren, zu bearbeiten oder anzuzeigen, und das vom Hersteller zur Verwendung durch Gesundheitsdienstleister bei der Patientenversorgung oder durch Patienten für den Zugang zu ihren elektronischen Gesundheitsdaten bestimmt ist
Diese „prioritären Kategorien personenbezogener elektronischer Gesundheitsdaten“ legt der Anhang I fest. Dazu zählen:
- „Patientenkurzakten“ inklusive demografischer Daten, Allergien, Anamnesen, Diagnosen, medizinischer und pflegerischer Verfahren (Behandlungen?) und Medikation
- Elektronische Verschreibungen und Abgabe von Arzneimitteln
- Medizinische Bildgebung und damit verbundene Befunde
- Ergebnisse elektronischer Untersuchungen wie Laboruntersuchungen einschließlich der zugehörigen Befunde
- Entlassungsberichte
Typische Beispiele für EHR-Systeme sind klinische Informationssysteme wie Krankenhaus-, Radiologie- und Laborinformationssysteme. EHR-Systeme sind auch von den Patienten verwendete Gesundheitsapps, wenn diese laut Zweckbestimmung Zugang zu den elektronischen Gesundheitsdaten ermöglichen.
c. Hersteller von Medizinprodukten und IVD
Medizinprodukte- und IVD-Hersteller sind dann betroffen, wenn sie behaupten, dass ihre Produkte mit den EHR-Systemen interoperabel seien. Das regelt Artikel 27 der EHDS-VO.
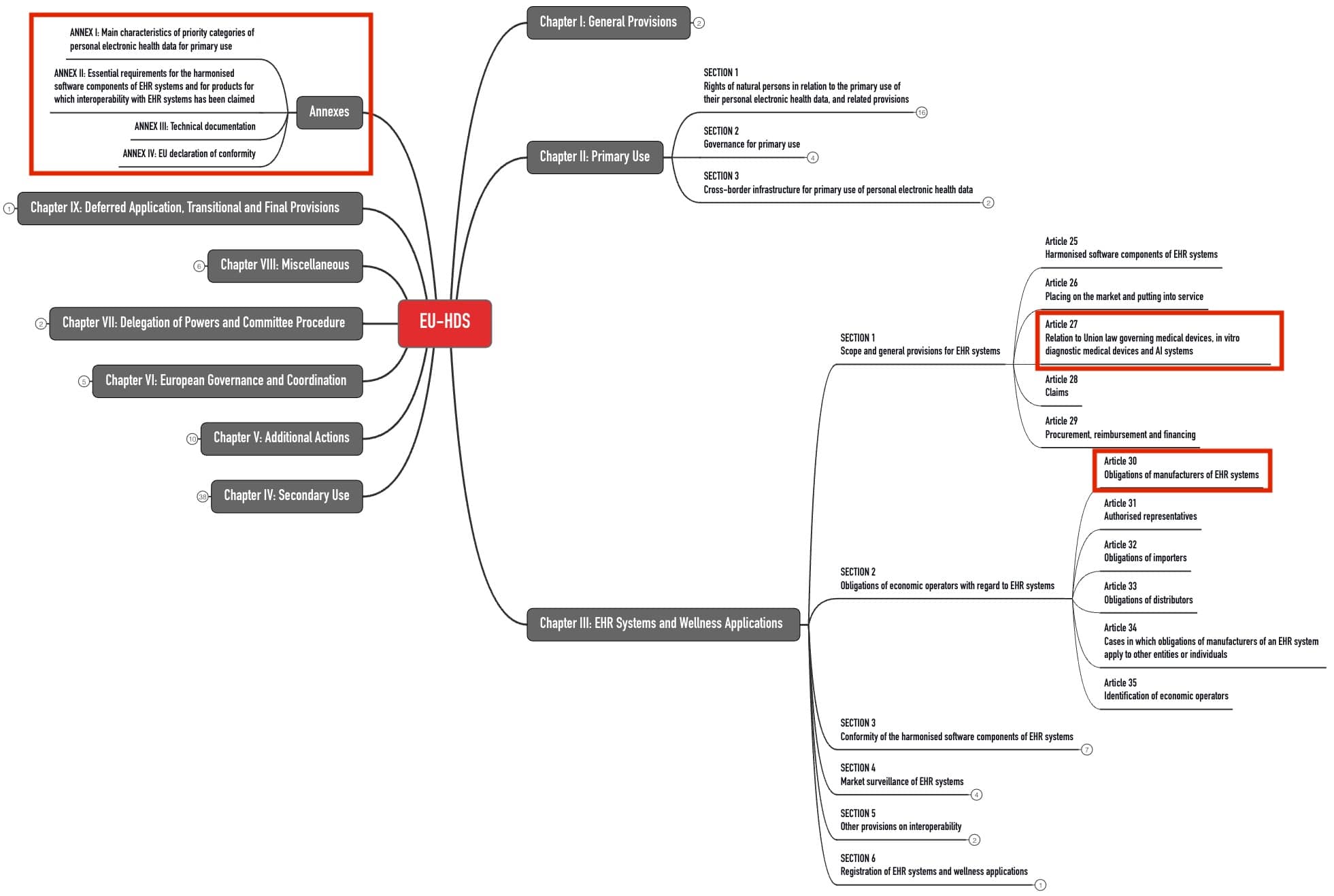
3. Pflichten der EHR-Hersteller
a. Interoperabilität der Produkte
Die oben genannten „prioritären Kategorien personenbezogener elektronischer Gesundheitsdaten“ müssen über ein maschinenlesbares Format übertragbar sein zwischen „verschiedenen Softwareanwendungen, Geräten und Gesundheitsdienstleistern“ (Artikel 15). Dafür sind Prüfverfahren geplant.
Für den grenzüberschreitenden Datenaustausch wird die Kommission eine zentrale Interoperabilitätsplattform (MyHealth@EU) einrichten.
Die EHR-Hersteller müssen in ihrer Software eine „europäische Interoperabilitätssoftwarekomponente“ integrieren.
eine Softwarekomponente des EHR-Systems, die personenbezogene elektronische Gesundheitsdaten einer prioritären Kategorie für Primärnutzung gemäß dieser Verordnung in dem europäischen Austauschformat für EHR gemäß dieser Verordnung bereitstellt und empfängt und die unabhängig ist von der europäischen Protokollierungssoftwarekomponente für EHR-Systeme;
EHDS-VO Artikel 2 (2) n)
Die Komponenten dieser Interoperabilitätssoftware müssen den Anforderungen des Anhangs II genügen, insbesondere dessen Abschnitt 2.
Dazu zählen die Anforderungen, dass die Softwarekomponente die Gesundheitsdaten im standardisierten Austauschformat senden und empfangen können muss und dabei keine zulässigen Zugriffe verbieten, einschränken oder erschweren darf.
b. Datenschutz und -sicherheit der Produkte
Zudem müssen die Produkte die Anforderungen an den Datenschutz und die IT-Sicherheit erfüllen. Die EHDS-VO ergänzt dazu die „Cybersecurity-Verordnung“ (EU) 2024/2847.
Diese verpflichtet die EHR-Hersteller, die grundlegenden Anforderungen der Cybersecurity-Verordnung (Anhang I) im Rahmen der Konformitätsbewertung nach EHDS-VO zu prüfen.
Die Anforderungen an den Datenschutz und die Datensicherheit bedingen, dass EHR-Systeme „zuverlässige Mechanismen zur Identifizierung und Authentifizierung“ bieten, was die EHDS-VO in Anhang II, Abschnitt 3.1 auch fordert.
Zudem verlangt die Verordnung eine „europäische Protokollierungssoftwarekomponente“.
eine Softwarekomponente des EHR-Systems, die protokollierte Informationen über den Zugang von Angehörigen der Gesundheitsberufe oder anderer Einzelpersonen zu prioritären Kategorien personenbezogener elektronischer Gesundheitsdaten gemäß der vorliegenden Verordnung in dem in Anhang II Nummer 3.2 dieser Verordnung festgelegten Format bereitstellt, und die unabhängig von der europäischen Interoperabilitätssoftwarekomponente für EHR-Systeme ist;
EHDS-VO Artikel 2 (2) o)
Diese Komponente protokolliert, wer wann auf welche Daten zugreift und woher die Daten stammen.
c. Weitere Anforderungen
Die weiteren Anforderungen an die Hersteller von EHR-Systemen dürften Medizinprodukteherstellern bekannt vorkommen:
| Anforderungen | Kommentare |
| Nachweis, dass die o. g. grundlegenden Anforderungen erfüllt sind | Hierzu plant die EU bis 26.03.2027 Common Specifications zu veröffentlichen. |
| Technische Dokumentation gemäß Artikel 37 und Anhang III | Im Vergleich dazu sind die Anforderungen an die TD von Medizinprodukten deutlich umfangreicher. |
| Konformitätserklärung | |
| CE-Kennzeichnung | |
| Registrierung | Sind diese Produkte auch Medizinprodukte oder IVD, wird die EUDAMED die Datenbank zur Registrierung sein. |
| Pflicht zu Korrekturmaßnahmen | Das setzt die Zusammenarbeit mit den Bevollmächtigten, Händlern und Behörden voraus. |
| Bereitstellen des Quellcodes in begründeten Fällen | Damit sollen Behörden die Einhaltung der grundlegenden Anforderungen prüfen können. |
Die Mitgliedsstaaten können zwar nationale Anforderungen an die Konformitätsbewertung festlegen (Artikel 41). Aber besondere Anforderungen an das Konformitätsbewertungsverfahren stellt die EHDS-VO nicht. Insbesondere sind keine Benannten Stellen für die Konformitätsbewertung von EHR-Systeme vorgesehen.
Die Konformitätsbewertungsverfahren nach anderen EU-Verordnungen (insbesondere MDR, IVDR, AI Act) bleiben unberührt.
4. Pflichten anderer Akteure
a. Pflichten der sonstigen „EHR-Wirtschaftsakteure“
Vergleichbar mit der MDR und IVDR sind die Pflichten an weitere Wirtschaftsakteure:
- Bevollmächtigte
- Importeure (hier „Einführer“ genannt)
- Händler
b. Pflichten der Hersteller von Medizinprodukten und IVD
Auch die Medizinprodukte- und IVD-Hersteller, die gemäß Artikel 27 von der EHDS-Verordnung betroffen sind, müssen die grundlegenden Anforderungen an die beiden Softwarekomponenten erfüllen:
- Europäische Interoperabilitätssoftwarekomponente für EHR-Systeme
- Europäische Protokollierungssoftwarekomponente für EHR-Systeme
Der Anhang II Abschnitt 2 der EHDS-Verordnung spezifiziert diese bereits oben beschriebenen grundlegenden Anforderungen.
Wie die Hersteller der EHR-Systeme, so müssen auch die Medizinprodukte- und IVD-Hersteller künftig gemeinsame Spezifikationen („Common Specifications“ CS) erfüllen, welche die Kommission bis zum 26.03.2027 erlassen will. Den „Scope“ dieser CS bestimmt der Artikel 36.
Ein zusätzliches Konformitätsbewertungsverfahren ist für diese Produkte nicht vorgesehen. Auch müssen diese Hersteller ihre Produkte in keiner weiteren Datenbank registrieren.
c. Pflichten der Hersteller von Wellness-Anwendungen
Auch die Hersteller von Wellness-Anwendungen fallen in den Anwendungsbereich der EHDS-VO, falls sie behaupten, dass ihre Produkte „mit einem EHR-System interoperabel“ seien.
jede vom Hersteller für die Verwendung durch eine natürliche Person bestimmte Software oder Kombination aus Hardware und Software für die Verarbeitung elektronischer Gesundheitsdaten, speziell für die Bereitstellung von Informationen über die Gesundheit natürlicher Personen oder für die Leistung von Pflege zu anderen Zwecken als der Gesundheitsversorgung.
Die Anforderungen an diese Hersteller sind im Vergleich zu den Anforderungen an die Medizinprodukte- und IVD-Hersteller geringer. Sie betreffen:
- Kennzeichnung dieser Anwendung (Artikel 47)
- Interoperabilität (Artikel 48)
- Registrierung (Artikel 49)
Eine (zusätzliche) CE-Kennzeichnung fordert die EHDS-VO nicht.
5. Tipps zur Umsetzung
a. Betroffenheit klären
Zuerst sollten Sie als Hersteller klären, ob Sie mit Ihren Produkten, die Sie im Markt haben oder in den Markt bringen wollen, unter den Anwendungsbereich der EHDS-VO fallen.
Dazu ist es notwendig, sich eine Übersicht über alle Entwicklungsprojekte zu verschaffen sowie alle im Markt befindlichen und alle geplanten Produkte zu „qualifizieren“. Das kann voraussetzen, dass Sie die Zweckbestimmung dieser Produkte präzisieren müssen.
b. Konsequenzen abschätzen
Zwar wirken die Anforderungen an die Hersteller (Medizinprodukte, IVD, EHR-Systeme) im Vergleich zu anderen Verordnungen bewältigbar. Allerdings betreffen diese Anforderungen stark die technische Implementierung der Systeme. Im ungünstigen Fall wird eine Neuentwicklung der Produkte notwendig.
Daher sollten Sie als Hersteller abschätzen, welche Ihrer Produkte Sie noch fit machen können, insbesondere um die Anforderungen an die Interoperabilität und Protokollierung zu erfüllen, und welche Aufwände dafür anfallen.
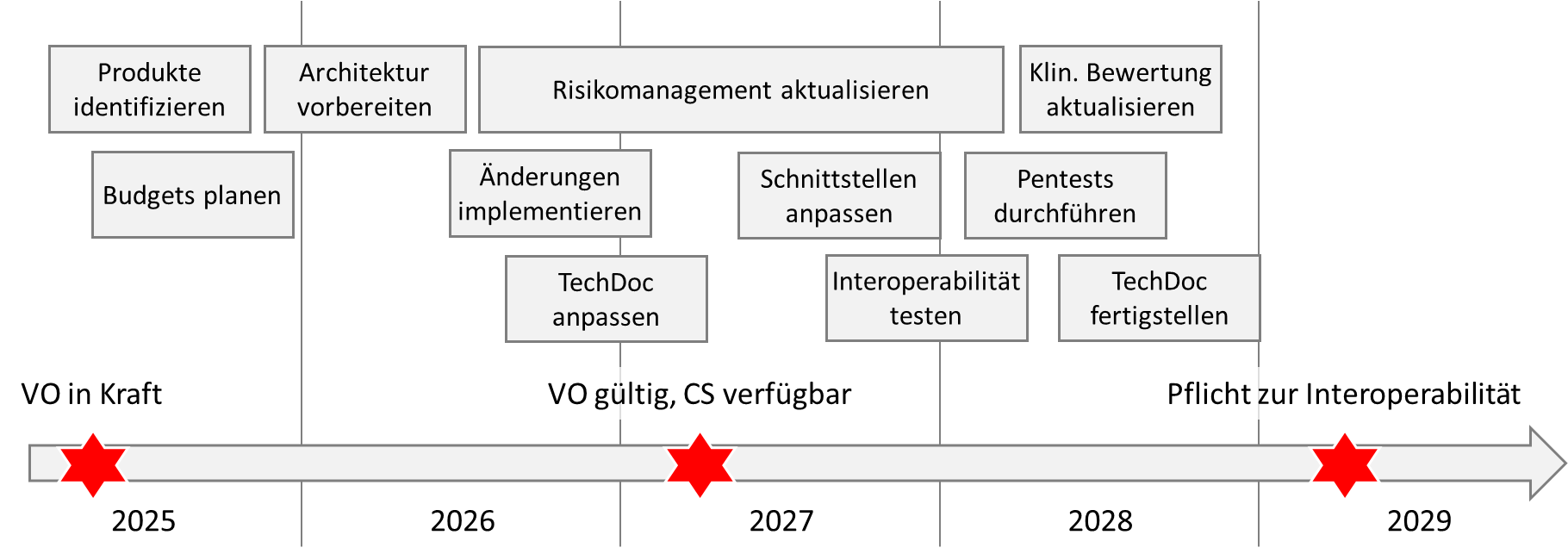
c. Übergangsfristen beachten
Vor der Planung sollten Sie die Übergangfristen beachten:
Die Verordnung ist am 26. März 2025 in Kraft getreten. Sie gilt ab dem 26. März 2027.
Sie gewährt Herstellern von EHR-Systemen sowie Medizinprodukten und IVD jedoch teilweisen Aufschub:
Sie müssen die Anforderungen an die Softwarekomponenten und damit an die Interoperabilität bezüglich der „prioritären Kategorien personenbezogener elektronischer Gesundheitsdaten“ erst 2029 erfüllen, bei Daten der medizinischen Bildgebung oder von medizinischen Untersuchungen (z.B. Labor) sogar erst 2031.
Auch für Inhouse-hergestellt EHR-Systeme gewährt die Verordnung eine Übergangsfrist bis 2031.
d. Projekte planen
Sind die Aufwände und der späteste Fertigstellungstermin bekannt, können Sie sich als Hersteller an die Planung machen. Dabei sollten Sie die Auswirkungen der Produktänderungen beachten:
- Überarbeitung der Datenschutz-, Rollen-, Authentifizierungs- und Autorisierungskonzepte
- Änderungen der Software-Architektur und Erweiterung um die geforderten Komponenten
- Analyse, wie sich die geänderten Schnittstellen auf die Interoperabilität mit anderen Produkten auswirken
- Ergänzung des Risikomanagements
- Erneute Durchführung von Pen-Tests
- Erweiterung der Post-Market Surveillance um weitere Datenquellen
Angesicht der Tatsache, dass viele Entwicklungsprojekte zwei und mehr Jahre dauern, kann ein zeitnaher Beginn der Umsetzung notwendig, schlimmstenfalls sogar bereits überfällig sein.
Warten Sie nicht auf die Common Specifications. Sie können sich nicht darauf verlassen, dass diese in der versprochenen Frist veröffentlicht werden. Zudem besteht die Gefahr, dass die Zeitspanne zwischen dem Fristende (März 2027) und der Pflicht zur Umsetzung (März 2029) zu kurz ist.
e. Zusammenfassung
Somit könnte Ihr Fahrplan wie in der folgenden Abbildung skizziert aussehen.
6. Fazit und Zusammenfassung
a. Nachvollziehbare Zielsetzung
Der Wunsch ist nachvollziehbar, in einem European Health Data Space Gesundheitsdaten strukturiert zu sammeln und nutzbar zu machen – sei es für die Patientenversorgung (primäre Nutzung) oder für Forschung, Statistik und politische Entscheidungsfindung (sekundäre Nutzung).
b. Gegebenenfalls herausfordernde Umsetzung
Sollten umfangreichere Änderungen der Produkte und deren Software-Systeme notwendig sein, dürften die Hersteller die Übergangsfristen nicht mehr als sehr großzügig empfinden. Denn die Aufwände sind hoch (s. o.).
Zudem steckt der Teufel auch hier im Detail:
- Es gibt derzeit keine Common Specification. Wenn diese erst 2027 erscheinen, bleiben den Herstellern nur noch zwei Jahre, diese zu erfüllen.
- Die Pflicht zur Protokollierung vieler Aktionen, zur Speicherung und Zugänglichmachung von Daten kann zu Konflikten mit den Anforderungen an den Datenschutz führen.
- Die Hersteller müssen genau überlegen, wie sie die Zweckbestimmung bzw. den bestimmungsgemäßen Gebrauch ihrer Produkte formulieren. Denn davon hängt die Anwendbarkeit der EHDS-Verordnung ab.
- Produkte können gleichzeitig als EHR-System, als Medizinprodukt und als Hochrisiko-KI-System zählen. Dann überlagern sich die Anforderungen von drei EU-Verordnungen.
- Das Ganze wird nur funktionieren, wenn auch die EU und nationale Behörden ihre sehr umfangreichen Hausaufgaben machen. Die Historie der EUDAMED und Common Specifications für die MDR bzw. IVDR wecken Skepsis.
c. Schnelle Klärung ratsam
Die oben genannten Tipps helfen, schnell Klarheit zu schaffen – auch darüber, wann mit der Umsetzung spätestens begonnen werden sollte. So gelingt es den Herstellern besser, ihre Entwicklungsprojekte zu planen und die notwendigen Ressourcen zu allokieren.
Klären Sie rasch, ob, und falls ja, wie Sie betroffen sind. Planen Sie dann das Projekt und die notwendigen Budgets. Wenn Sie dabei Hilfe benötigen, dann schreiben Sie mir eine kurze E-Mail über info[at]johner-institut.de.
Das Johner Institut unterstützt die Hersteller auch bei der Umsetzung der Verordnung 2025/327:
- Abschätzung, welche Produkte von der Verordnung betroffen sind
- Bewertung der Software-Architektur und Auswirkungen der Änderungen
- Festlegung eines Umsetzungsplans
- Aktualisierung des Risikomanagements und der klinischen Bewertung
- Bewertung der IT-Sicherheit einschließlich Pen-Testing
Änderungshistorie
- 2025-08-14: Im Abschnitt 1. b. die Artikelnummerierung korrigiert. Es wurde fälschlicherweise der Artikel 51 statt 53 referenziert
- 2025-04-08: Erste Version des Artikels veröffentlicht



{kind=link}
Im Artikelabschnitt „b. Vorteile für Hersteller von Medizinprodukten und IVD“ wird auf Artikel 51 verwiesen, die entsprechenden Stellen finden sich jedoch in Artikel 53. Vielen Dank für den sehr informativen Inhalt!
Danke, lieber Herr Goetz, für Ihren wertvollen Hinweis!
Ich habe die Stelle angepasst. Ich war in der Tat in der Nummerierung verrutscht.
Nochmals vielen Dank!
Beste Grüße, Christian Johner