
Mit dem eStar-Programm möchte die FDA die Effizienz von Zulassungsverfahren (z. B. der 510(k)-Verfahren) durch Digitalisierung erhöhen. Bei der Weiterentwicklung von eStar arbeitet das Johner Institut mit der FDA zusammen.
Wie zukunftsweisend dieser Ansatz ist und ob Sie daran teilnehmen sollten oder gar müssen, erfahren Sie in diesem Artikel.
1. Was ist das eStar-Programm?
Das eStar-Programm ermöglicht Medizinprodukteherstellern, ihre Zulassungsunterlagen über ein interaktives PDF bei der FDA einzureichen.
Die FDA schreibt in ihrem finalisierten Guidance-Dokument Electronic Submission Template for Medical Device 510(k) Submissions, dass sie seit dem 01.10.2023 510(k)-Einreichungen nur noch im eSTAR-Format und per Upload über das FDA-Portal akzeptiert.
Ähnliches gilt für De-Novo-Einreichungen. Diese werden gemäß dem Guidance-Dokument Electronic Submission Template for Medical Device De Novo Requests ab dem 01.10.2025 ebenfalls nur noch im eSTAR-Format akzeptiert.
a) Interaktivität
Die Interaktivität hat mehrere Aspekte.
Integrierte Fehlerprüfung
Das PDF markiert Bereiche, in denen Daten offensichtlich noch Fehler enthalten, in Rot, und korrekte Bereiche in Grün (s. Abb. 1).
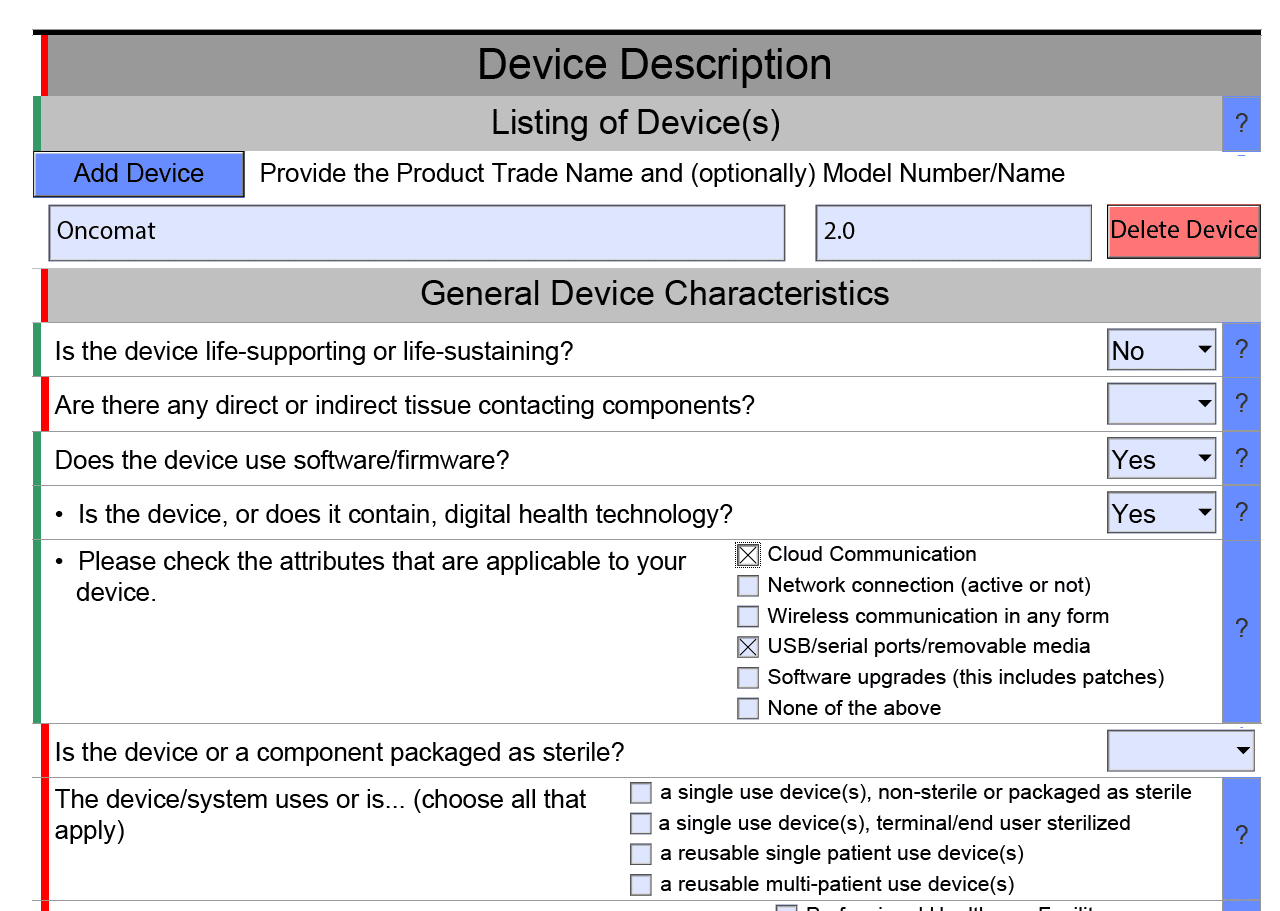
Automatisiertes Ein- und Ausblenden von Bereichen
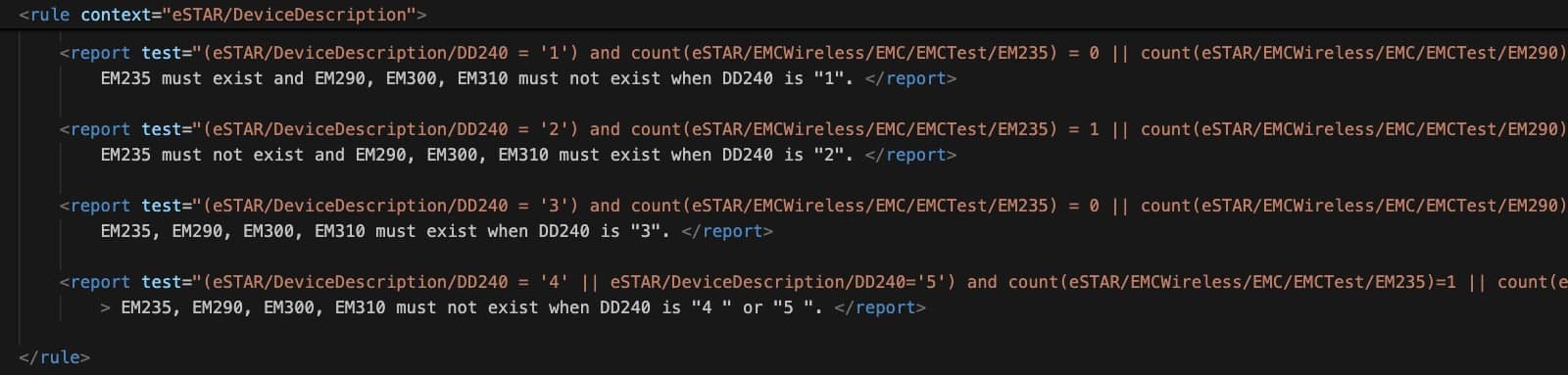
Das interaktive Dokument blendet Bereiche ein oder aus, abhängig von der Auswahl. Beispielsweise zeigt das Dokument automatisch Details zu den Software-Unterlagen, wenn man auswählt, dass das Produkt Software enthält oder eine Standalone-Software ist (s. Abb. 1). Diese Logik zum Ein- und Ausblenden legt die FDA in Schematron-Dateien fest (s. Abb. 2).
Integration von Anhängen
Das PDF erlaubt, Anhänge beizufügen. Aus Sicht der FDA müssen diese Anhänge sogar beigefügt werden. Dabei gibt es keine Einschränkung des Datenformats. Einzig die Größe des resultierenden PDF-Dokuments ist auf 1 GB beschränkt (s. Abb. 3).

Möglichkeiten zum Import und Export
Alle Daten, mit Ausnahme der Anhänge, können über das PDF aus XML importiert und nach XML exportiert werden.
b) Elektronische Einreichung
Inzwischen erfolgt die Einreichung nicht mehr auf einem physischen Datenträger, sondern elektronisch. Abhängig vom zuständigen FDA Center reichen Hersteller über das CDRH Portal ein oder über das ESG Gateway bei CBER-bezogenen Einreichungen.
c) eStar versus eCopy
Tatsächlich stellt das eStar-Programm nach dem eCopy/eSubmitter-Programm die nächste Evolutionsstufe in der Digitalisierung dar. Bei dem eCopy/eSubmitter-Programm gab die FDA „nur“ die Formatierung der elektronischen Einreichungsakte vor, an die sich die Hersteller halten müssen. Eine vorgegebene Struktur der Dokumente selbst oder gar strukturierte Daten wie beim eStar-Programm gibt es beim eCopy/eSubmitter-Programm nicht.
Wenn Sie mehr zur Geschichte dieser beiden Programme wissen wollen und einen Vergleich wünschen, empfiehlt sich ein Blick in dieses Guidance-Dokument.
2. Wer kann oder darf das eStar-Programm nutzen?
Offensichtlich soll das eStar-Programm die Medizinproduktehersteller dabei unterstützen, Zulassungsunterlagen schneller und in besserer Güte zusammenzustellen und einzureichen. Doch es wendet sich nicht an alle Hersteller und eignet sich nicht für alle Zulassungsverfahren.
Das eStar-Programm ist nur anwendbar für:
- 510(k)-Zulassungsverfahren (traditional, abbreviated, special)
- De-Novo-Zulassungsverfahren
- Pre-Submissions
- Medizinprodukte, IVD und Kombinationsprodukte
- Gewisse PMA-Zulassungsverfahren (Original, PMA Panel Track Supplements, PMA Real-Time, PMA 180 Day Supplements)
Die Teilnahme am eStar-Programm ist explizit freiwillig. Hersteller und Produkte, für die das Verfahren anwendbar ist, dürfen, aber müssen nicht daran teilnehmen.
Ausgenommen sind 510(k)-Verfahren. Hier ist die Teilnahme am eStar-Programm Pflicht.
3. Wie hilft das eStar-Programm?
Das eStar-Programm nützt den Herstellern ebenso wie der FDA.
a) Unterlagen in besserer Güte
Die FDA erhält die Unterlagen in höherer Güte, weil das PDF den Herstellern hilft, offensichtliche Fehler wie fehlende Informationen oder Tippfehler (z. B. bei Produkt-Codes) zu vermeiden. Dazu tragen beispielsweise Drop-Down-Listen und Überprüfungen der Inhalte auf Vollständigkeit und Konsistenz bei.
b) Mehr Sicherheit
Das PDF verifiziert markiert unvollständige oder fehlende Informationen sowohl auf Ebene des einzelnen Datums als auch auf Ebene des gesamten Dokuments (s. Abb. 3).

c) Geringerer Aufwand
Die Hersteller und die FDA ersparen sich die RTA-Korrekturschleife. Der Rest des Prozesses bleibt allerdings unverändert:
The remainder of a 510(k) review will be conducted according to the FDA guidance, „The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)],” following the procedures identified in 21 CFR 807 subpart E. A De Novo review will be conducted according to the FDA guidance, “De Novo Classification Process (Evaluation of Automatic Class III Designation),” following the procedures identified in 21 CFR 860, subpart D.
Die FDA muss keine Inhalte per Copy-and-paste in die eigenen Systeme übergeben. Auch das erspart Aufwand.
Zudem findet die Behörde die Unterlagen schneller, weil der Zugriff auf die Inhalte und Anhänge direkt über das PDF-Dokument möglich ist und weil die Hersteller gezwungen werden, die Anhänge in der von der FDA vorgegebenen Struktur zusammenzustellen. Auch dies erspart der Behörde Arbeit. Sie sagt jedoch keine kürzeren Bearbeitungsdauern zu.
4. Bewertung des eStar-Programms
a) Positiv
Die FDA unterstützt mit diesem Programm die Hersteller dabei, vollständige, konsistente und korrekte Unterlagen zusammenzustellen.
Indem sie viele Vorprüfungen automatisiert, profitiert auch die Behörde selbst. Das wiederum kommt den Herstellern zugute, weil Zulassungsprozesse schneller ablaufen und insbesondere unnötige Iterationsschleifen (inkl. RTA-Prüfung) vermieden werden können.
Die Struktur des PDF-Dokuments (s. Abb. 4) und die Aufteilung der Anhänge gibt den Herstellern wertvolle Anregungen, um die eigenen Unterlagen zu strukturieren. Das gilt sowohl auf Ebene der Verzeichnisse als auch für die Dokumente selbst.

b) Kritik
Die Möglichkeit eines XML-Imports kann bestenfalls eine Übergangstechnologie sein, um die Informationen aus bestehenden Systemen in das PDF-Dokument der FDA zu übertragen. Das ist zwar besser als ein manuelles Copy-and-paste, aber kein Ersatz für eine API.
Die FDA gibt die (Struktur der) erwarteten Anhänge vor. Damit zwingt sie die Hersteller, ihre Dokumente FDA-spezifisch zusammenzustellen. Dies führt zu den üblichen Fleißaufgaben von Regulatory Affairs Managern und Managerinnen. Aufgaben, die eine vollständige Digitalisierung der Prozesse überflüssig machen könnte und sollte.
c) Bewertung und Ausblick
Die FDA ist mit dem eStar-Programm einen deutlichen Schritt weiter als viele Benannte Stellen mit ihren „Application Forms“. Diese können oft weder von der Funktionalität noch von der Gebrauchstauglichkeit und Interoperabilität (XML-Import und Expert) mithalten.
Das interaktive PDF ist eine Software, die wie jede andere Software validiert und weiterentwickelt werden muss. Bisher erfolgt die Aktualisierung durch die FDA sehr zeitnah. Zum Beispiel ist das neue Guidance-Dokument zu Software mit der Unterscheidung nach „Basic Documentation“ und „Enhanced Documentation“ inzwischen umgesetzt.
d) Vergleich mit der DZP
Das eStar-Programm ist ein Schritt in die richtige Richtung. Gleichzeitig wird es diesen unvollständigen Digitalisierungsgrad auf Jahre hin zementieren.
Wünschenswert wäre gewesen, wenn die FDA bereits den gleichen Digitalisierungsgrad wie die digitale Zulassungsplattform (DZP) des Johner Instituts erreicht hätte:
- (Weitgehend) vollständige Erfassung und Prüfung strukturierter Daten statt Erfassung von Anhängen und Textfeldern, die nicht computerauswertbar sind. So nutzt eStar ein Textfeld für die komplette Zweckbestimmung inklusive Charakterisierung der Nutzer und Patienten.
- (Fast) dokumentenfreie Einreichung
- Weitgehend vollständige automatisierte Prüfung aller Daten
- Kompletter Zugriff auf alle Daten via API und damit Integration in bestehende Systeme der Hersteller und Behörden bzw. Benannten Stellen
- Keine Medienbrüche und insbesondere keine Notwendigkeit, Informationen in Dokumente (Papier, PDF) zu überführen oder gar über physische Datenträger zu verschicken
- Unterstützung des gesamten Prozesses bis zur erfolgreichen oder endgültigen Ablehnung der Zulassung einschließlich des Feedbacks durch die Behörde bzw. Benannte Stelle
5. Fazit
a) Zusammenfassung
Mit dem eStar-Programm geht die FDA den nächsten Schritt bei der Digitalisierung der Zulassungsprozesse. Sie treibt damit das auf die Spitze, was man mit einem dokumenten- und PDF-basierten Ansatz erreichen kann.
Die Interaktivität der PDF-Dokumente mit integrierter Anwendungslogik und einer Mischung aus strukturierten Daten und Anhängen lässt erahnen, was mit einer vollständig automatisierten Erfassung und Prüfung von Zulassungsdaten möglich wäre.
Bis dahin müssen die Hersteller mit Dokumenten arbeiten und dabei die folgenden Leitlinien beachten:
- Das bestehende Guidance-Dokument „Providing Regulatory Submissions for Medical Devices in Electronic Format — Submissions Under Section 745A(b) of the Federal Food, Drug, and Cosmetic Act”, auch wenn dieses nicht mehr ganz aktuell ist, da es vor dem eStar-Programm publiziert wurde und dieses noch nicht enthält.
- Wichtiger ist das neue Guidance-Dokument „Electronic Submission Template for Medical Device 510(k) Submissions“ und das Guidance-Dokument Electronic Submission Template for Medical Device De Novo Requests.
Das Johner Institut unterstützt Hersteller und Benannte Stellen bei der digitalen Transformation. Interesse? Dann nehmen Sie gleich Kontakt auf!
Versionshistorie:
- 2024-10-15: Hinweis zur verpflichtenden Nutzung von eSTAR für De Novo Requests ab dem 01.10.2025 ergänzt
- 2024-08-20: Bewertungen in das neue Kapitel 4 verschoben. Nummerierung konsequent umgesetzt. Einleitung ergänzt. Kapitel 1.a) neu strukturiert und um Bild der Schematron-Datei (Abb. 2) ergänzt.
- 2023-09-15: Hinweise zur elektronischen Einreichung und Verfügbarkeit von eSTAR für Kombinationsprodukte ergänzt
- 2022-09-23: Hinweise zum neuen eSTAR Guidance Dokument ergänzt


Hallo liebes Johner Team,
haben Sie Vorgaben gefunden, wie PDFs, welche man als Attachment an das EStar pdf hängen soll, aussehen müssen?
Vielen Dank!
Guten Tag Frau Berndt,
Hinweise zu den Anhängen zum eSTAR finden Sie direkt im Template. Konkrete Vorgaben zu angehängten PDFs gibt es allerdings nicht. Die FDA schreibt:
„eSTAR will prevent unacceptable attachment types from being added. We tested eSTAR submissions up to 1GB in size. If your eSTAR is greater than 1GB, file handling may be delayed. Large attachments will increase save times.“
oder
„Please combine attachments of similar content (for example, Software Requirements Specifications) when possible so that only one attachment needs to be provided to each attachment type question in the eSTAR. Attachments may be combined in Adobe Acrobat Pro by choosing „Tools“ then „Combine Files.“ We recommend that combined documents have bookmarks or contain a Table of Contents for easier review.“
Herzliche Grüße
Luca Salvatore
Dear Mr. Salvatore,
Thanks for insightful article.
I have two questions here, firstly for most of sections in eSTAR there is a passage with box underneath asking for applicable general and special control for theatre particular section for chosen device product code, what is exactly being asked there? If there is no special control guidance is available but the product code regulation only refers to performance standards as special control, how one should take the lead in answering such parts?
Secondly for the reference section in eSTAR do we have to refer all the literature data that is used for product development? Starting from literature on choice of materials of device, patient population and biocompatibility aspect etc?
Best,
Rabel
Dear Rabel,
you should identify for each applicable section (e.g. sterility, performance testing,…) if you have applied any kind of FDA special controls applicable to your device. This could be a device-specific „special control guidance“, a performance standard, specific labeling requirements, etc. If in your case there is an applicable performance standard, you should identify it in the respective special control section of the eSTAR.
Regarding the literature: You should only refer to literature which you use to demonstrate substantial equivalence/ safety and effectiveness of your device. This could be literature related to clinical data or liteature on the evaluation of biocompatibility of materials used.
Best regards
Luca
Hallo,
was ist die digitale Zulassungsplattform (DZP)? Finde dazu nichts auf eurer Website.
beste Grüße
Guten Tag,
die Digitale Zulassungsplattform ist Bestandteil des Realtime Compliance Systems: https://www.johner-institut.de/digitale-dienstleistungen/realtime-compliance-system/. Ziel ist die digitale Transformation der Zulassungsprozesse, Ende-zu-Ende (d.h. von Herstellersystemen bis hin zu Benannten Stellen/Behörden).
Wir begleiten u.a. Hersteller im Rahmen des Fit-for-Future Programms (https://www.johner-institut.de/dt/fit-for-future-programm/) dabei, ihre regulatorischen Prozesse zu digitalisieren. Dies erfolgt unter Nutzung der digitalen Zulassungsplattform.
Freundliche Grüße
Luca Salvatore