510(k)-Verfahren, auch “Premarket Notification” genannt, gehören zu den gängigsten Verfahren, nach denen in den USA Medizinprodukte zugelassen werden. Das Konzept basiert darauf, die Äquivalenz mit einem Vergleichsprodukt nachzuweisen. Doch dabei passieren schnell die immer gleichen Fehler, an denen die gesamte 510(k)-Zulassung scheitern kann.
Das muss nicht sein! Erfahren Sie in diesem Beitrag, welches die fünf häufigsten Fehler bei 510(k)s sind und wie Sie diese vermeiden.
Hier erfahren Sie, wie das 510(k)-Verfahren funktioniert:
- FDA 510(k): Premarket Submission
- Special 510(k): Wann die FDA diese „Abkürzung“ erlaubt
- Abbreviated 510(k) – Wann die Abkürzung erlaubt ist
sowie im Auditgarant.

1. Fehler: Product Code falsch identifiziert
Anders als im EU-Recht, wo Produkte nach Regeln von Fall zu Fall klassifiziert werden, gibt es in den USA bereits feststehende Codes für bestimmte Produktklassen. Hersteller müssen also “nur” den für ihr Produkt passenden Code identifizieren.
Der Product Code liefert wertvolle Informationen für die Einreichung, unter anderem:
- Den geforderten Submission Type (z. B. 510(k), 510(k) Exempt)
- Produktklasse (I, II oder III)
- Regulation Number mit der Produktbeschreibung
- Recognized Consensus Standards zum Produkt
- Anforderungen an die Special Controls
Es gibt allerdings mehr als 1700 Product Codes! Deshalb ist die Identifikation des richtigen Produkt Codes eine häufige Fehlerquelle.
Lösung:
Der Zeitpunkt ist entscheidend: Den Product Code sollten Sie ermitteln, wenn die Zweckbestimmung (Intended Use) Ihres Produkts festgelegt ist.
Die FDA beschreibt den Begriff „intended use“ wie folgt:
intended use means the general purpose of the device or its function, and encompasses the indications for use.
Wir verwenden im Folgenden den Begriff „Zweckbestimmung“ in diesem Sinne.
Die Schritte sind:
- Schritt 1: Zweckbestimmung festlegen
- Schritt 2: Auf dem Markt befindliche Vergleichsprodukte suchen und in der 510(k)-Datenbank deren Product Codes herausfinden
- Schritt 3: In der FDA-Datenbank selbst nach Product Codes über Panels suchen
- Schritt 4: Gefundene Product Codes nach den passendsten Merkmalen bewerten und passenden Product Code festlegen
Das Johner Institut hilft Ihnen gern, falls Sie Fragen zum 510(k)-Verfahren haben. Natürlich beantworten unsere Expert:innen auch alle Fragen zu Product Codes. Kontaktieren Sie uns!
Product Code durch “Classify Your Medical Device”
Auch die FDA bietet Hilfe an. Die Website Classify Your Medical Device beschreibt verschiedene Möglichkeiten, die Klassifizierung und somit den Product Code des jeweiligen Produkts zu ermitteln.
Bei Fragen: FDA kontaktieren
Bei Unsicherheiten können Sie die FDA direkt kontaktieren, und zwar
- unverbindlich per E-Mail über: DICE@fda.hhs.gov bzw. DeviceDetermination@fda.hhs.gov oder
- formell durch einen 513(g)-Request. Hierbei bittet der Hersteller um eine formale Stellungnahme bzw. Festlegung der Klassifizierung durch die FDA für das eigene Produkt.
Insbesondere ein 513(g)-Request verhindert, dass ein Produkt versehentlich mit einem falschen Code eingereicht wird. Allerdings erhebt die FDA hierfür Gebühren.
Die User Fee für den 513(g)-Request beträgt
- normal US$ 5.061,
- für kleine Unternehmen US$ 2.530.
Informationen darüber, wie ein 513(g) gestellt wird, sind in folgendem Guidance Dokument enthalten: FDA and Industry Procedures for Section 513(g) Requests for Information under the Federal Food, Drug, and Cosmetic Act.
2. Fehler: Produktspezifische Anforderungen nicht beachtet
Die FDA hat zu zahlreichen Produkten spezifische Anforderungen festgelegt, die eingehalten und im 510(k) nachgewiesen werden müssen. Unwissenheit über diese Anforderungen oder deren unzureichende Berücksichtigung führen häufig zu Rückfragen der FDA. Beispielsweise
- werden produktspezifische Performance Standards nicht berücksichtigt,
- wird unzureichend begründet, warum bestimmte Standards nicht oder nur teilweise zutreffend sind,
- werden keine ausreichenden Produkttests durchgeführt,
- werden Special Control Guidelines nicht berücksichtigt oder
- die Declaration of Conformity ist fehlerhaft bzw. enthält nicht die entsprechenden anerkannten Normen.
Lösung:
Zunächst sollten Sie prüfen, ob es bestimmte produktspezifische Anforderungen gibt, die in einer Premarket Notification berücksichtigt werden müssen. Diese Anforderungen finden Sie heraus, indem Sie
- Ihr Produkt klassifizieren,
- die Special Control Guidelines der FDA durchgehen,
- in der Datenbank der FDA Guidance Documents nach dem jeweiligen Produkt suchen (so lassen sich etwaige Guidance-Dokumente mit Angaben über produktspezifische Anforderungen identifizieren.),
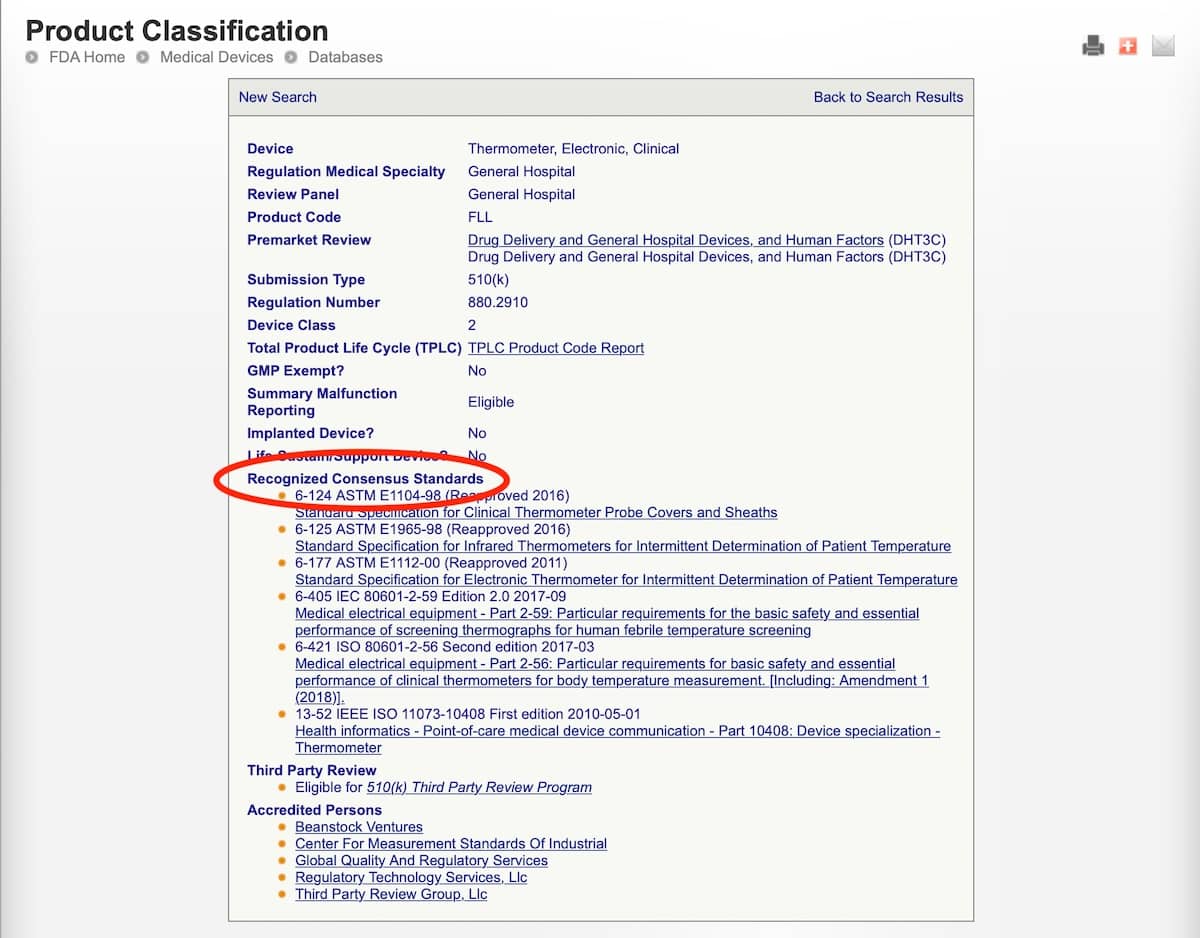
- Sie die FDA Product Classification Database durchsuchen. (So lassen sich etwaige Recognized Consensus Standards zum zugehörigen Product Code identifizieren.)
Beispiel:
3. Fehler: Predicate Device ist NSE (not substantially equivalent)
Beachten Sie den Fachartikel zu den Predicate Devices und der „Substantial Equivalence“.
Ein 510(k) basiert auf dem Konzept der “Substantial Equivalence” (SE). Das bedeutet, dass Hersteller nachweisen, dass das eigene Produkt genauso sicher oder sogar sicherer ist als ein äquivalentes, bereits legal auf dem Markt verfügbares Produkt. Ein Produkt ist äquivalent (SE), wenn es
- die gleiche übergeordnete Zweckbestimmung und
- gleiche technologische Eigenschaften hat (z. B. Material, Design, Energiequelle, Features).
Hat das Produkt die gleiche Zweckbestimmung, aber andere technologische Eigenschaften, muss anhand von Produktprüfungen, klinischen oder wissenschaftlichen Daten nachgewiesen werden, dass
- das Produkt ebenso sicher und wirksam ist wie das Vergleichsprodukt und
- es keine zusätzlichen Fragen bezüglich der Sicherheit und Wirksamkeit aufwirft.
Die Diskussion der Substantial Equivalence (SE-Diskussion) ist daher ein wichtiger Punkt einer Premarket Notification. Sie beinhaltet die Produktcharakteristiken des neuen Produkts und eine Erläuterung des Herstellers, inwiefern diese mit dem Äquivalenzprodukt vergleichbar sind.
Häufige Fehler, die dabei auftreten sind:
- Die FDA erkennt die Äquivalenz des Vergleichsprodukts (Predicate Device) nicht an, da die Substantial Equivalence nicht ausreichend nachgewiesen oder nicht ausreichend begründet wird. Eine Aussage wie „The device does not raise different questions of safety and effectiveness“ muss belegt werden.
- Fehlerhafte Verwendung von mehreren Vergleichsprodukten (Predicate Devices)
Die FDA erlaubt zwar die Nutzung von mehreren Vergleichsprodukten, z. B. wenn ein Produkt die Eigenschaften von zwei Vergleichsprodukten vereint, jedoch müssen alle Produkte über die gleiche Zweckbestimmung verfügen.
Beispiel: Ein Patientenmonitor, der verschiedene Parameter anzeigt
Zusätzlich müssen die technologischen Eigenschaften vergleichbar sein. Es ist nicht möglich, die Zweckbestimmung des einen und die technologischen Eigenschaften des anderen Produkts für den Substantial-Equivalence-Vergleich heranzuziehen.
Lösung:
Diese Fehler lassen sich mit folgenden Maßnahmen verhindern:
- Legen Sie ein geeignetes Predicate Device fest
Das Vergleichsprodukt muss dem eigenen Produkt in Bezug auf die Zweckbestimmung (Intended Use) und die technologischen Eigenschaften ähneln. Mehrere Vergleichsprodukte sind möglich, allerdings empfiehlt die FDA, ein primäres Predicate Device festzulegen.
Die folgenden Fragen helfen bei der Identifizierung eines geeigneten Predicate Devices:
- Hat mein Vergleichsprodukt die gleiche Zweckbestimmung?
- Hat es die gleichen technologischen Eigenschaften (z. B. mit Blick auf Design, Material, Features, Energiequelle)?
- Wenn das Produkt unterschiedliche technologische Eigenschaften hat, werfen diese zusätzliche Fragen bezüglich der Sicherheit und Wirksamkeit des Produkts auf?
Anhaltspunkte liefert auch der Entscheidungsbaum in Anhang A der FDA Guidance The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)].
- Führen Sie die Diskussion der Substantial Equivalence anhand von FDA-Guidance durch
Die Anforderungen an die SE-Diskussion sowie Hinweise, was bezüglich der Zweckbestimmung und technologischen Eigenschaften zu beachten ist, hat die FDA im Guidance-Dokument The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)] festgehalten.
4. Fehler: Formfehler
Formfehler gehören sicherlich zu den häufigsten Problemen bei aller Art von Anträgen. Daher prüft die FDA die formalen Anforderungen in zwei Schritten, bevor der 510(k) überhaupt einem Fachprüfer vorgelegt wird:
- Log-in und Acknowledgement Procedure: Hierfür muss die Premarket Notification die eCopy-Anforderungen einhalten und der User Fee muss bezahlt sein.
- Acceptance Review: Hierbei prüft die Behörde anhand der RTA (Refuse to Accept)-Checkliste, ob die Einreichung formal vollständig ist, bevor sie für das Substantive Review an den Fachprüfer weitergeleitet wird.
Bei 510(k)s gehören zu den häufigsten Formfehlern:
- Der Antrag erfüllt nicht die Anforderungen an das elektronische Einreichungsformat (eCopy).
- Das Anschreiben (Cover Letter) enthält nicht alle geforderten Informationen.
- Die User Fee wurde (noch) nicht bezahlt.
- Es fehlen Informationen, da z. B. die RTA-Checkliste nicht ausreichend berücksichtigt wurde.
Lösung:
- Klären Sie, welcher 510(k)-Typ in Frage kommt (special, abbreviated, traditional) und übermitteln Sie alle geforderten Informationen.
- Falls Sie unsicher sind, welche Informationen Sie an die FDA übermitteln müssen, können Sie einen sogenannten Pre-Submission Request stellen und Ihre Fragen direkt mit der FDA besprechen.
- Die Anforderungen an die Formatierung der elektronischen Einreichung finden Sie in den zugehörigen eCopy-Guidances.
- Außerdem sollten Sie vor Einreichung unbedingt die RTA-Checkliste der FDA durchgehen.
Hilfreich sind zudem die von der FDA zur Verfügung gestellten Tools:
- eSubmitter-eCopies Tool: Mit diesem Tool können eCopy-konforme Einreichungen erstellt werden.
- eCopy Validation Module: Dieses Tool ermöglicht es, eine Einreichung auf Konformität mit den eCopy-Anforderungen zu prüfen.
- eSTAR: Ein interaktives PDF der FDA, das Sie durch die Zusammenstellung eines 510(k) leitet. Inhalt und Struktur sind vorgegeben. Der Vorteil von eSTAR ist, dass die RTA (Refuse to Accept)-Prüfung entfällt.
5. Fehler: FDA Guidance-Dokumente nicht beachtet
Die FDA selbst bietet umfangreiche Hilfestellungen für 510(k)s an. Wer diese nicht oder nicht ausreichend beachtet, macht oft folgende Fehler:
- Wichtige Dokumente fehlen im Antrag.
- Die Einreichung ist so unübersichtlich gestaltet, dass der Prüfer Informationen nicht findet.
- Die Dokumente enthalten nicht alle von der FDA geforderten Informationen (z. B. unvollständige SE-Diskussion, unzureichende Risikoanalyse, unvollständige Software-Architektur).
- Die Informationen in der Einreichung sind nicht einheitlich oder widersprechen sich (z. B. unterschiedliche Zweckbestimmung an unterschiedlichen Stellen der Einreichung).
- Die Produktbeschreibung ist unvollständig und passt nicht vollständig zum identifizierten Produktcode.
Lösung:
Die FDA hat Guidance-Dokumente zu den geforderten Inhalten einer Premarket Notification veröffentlicht. Diese enthalten u. a. Listen mit den Dokumenten, die eingereicht werden müssen, und sollten bereits während des Entwicklungsstadiums berücksichtigt werden. So lässt sich sicherstellen, dass alle nötigen Dokumente erstellt werden.
Besonders beachtenswert sind die folgenden Dokumente:
- Format for Traditional and Abbreviated 510(k)s
- Für Produkte, die Software enthalten: Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices und das Draft-Dokument Content of Premarket Submissions for Device Software Functions
- Für Gebrauchstauglichkeit: Applying Human Factors and Usability Engineering to Medical Devices
Zahlreiche weitere Guidance-Dokumente zu verschiedenen Themen gibt es in der FDA-Database.
Zusammenfassung
510(k)s, die sogenannte Premarket Notification, sind für viele Produkte der vorgeschriebene Weg, um auf den amerikanischen Markt zu kommen. Vermeiden Sie jedoch die häufigsten Fehler und achten Sie auf folgende Punkte:
- Product Code richtig identifizieren
- Produkt klassifizieren über die Panels bzw. die Website Classify Your Medical Device
- Bei Unsicherheiten die FDA kontaktieren, entweder per E-Mail an DICE@fda.hhs.gov bzw. DeviceDetermination@fda.hhs.gov oder 513(g)-Request
- Produktspezifische Anforderungen bestimmen
- Nach der Klassifizierung die Special Control Guidelines der FDA durchgehen
- Die Datenbank der FDA Guidance Dokumenten nach dem jeweiligen Produktcode durchsuchen; dabei Recognized Consensus Standards mit Angaben über produktspezifische Anforderungen identifizieren
- Predicate Device ist NSE (not substantially equivalent)
- Geeignetes Predicate Device festlegen
- SE-Diskussion anhand von FDA-Guidance durchführen
- Form einhalten
- RTA-Checkliste durchgehen
- FDA-Tools nutzen: eSubmitter-eCopies Tool (eCopy-konforme Einreichungen erstellen), eCopy Validation Module (auf Konformität mit den eCopy Anforderungen prüfen), eSTAR (interaktive PDFs)
- Guidance Dokumente in der FDA-Database beachten
Mit der FDA-Serie im Auditgarant können Sie Ihre 510(k)-Einreichung so erstellen, dass Sie problemlos durch das Verfahren gelangen. Beachten Sie hierzu auch unsere Fachbeiträge zu 510(k)s.