
Mangelhaft gestaltete Gebrauchsanweisungen sind häufig Ursache für Benutzungsfehler, welche zu Schäden bei Patienten und Anwendern führen können. Zudem unterliegen Gebrauchsanweisungen für Medizinprodukte und IVD strengen regulatorischen Anforderungen. MDR, IVDR, die FDA sowie zahlreiche Normen stellen spezifische Anforderungen an Gebrauchsanweisungen.
In diesem Artikel erfahren Sie, wie Sie eine Gebrauchsanweisung schreiben,
- die hilft, Schäden bei Patienten, Anwendern oder Dritten zu vermeiden,
- die alle relevanten Regularien erfüllt und somit im Audit besteht,
- die nutzerfreundlich ist und dadurch die Zufriedenheit Ihrer Kunden erhöht und den Markterfolg steigert.
1. Definition und Synonyme
„bezeichnet vom Hersteller zur Verfügung gestellte Informationen, in denen der Anwender über die Zweckbestimmung und korrekte Verwendung eines Produkts sowie über eventuell zu ergreifende Vorsichtsmaßnahmen unterrichtet wird;“
“package insert; portion of the accompanying information that is essential for the safe and effective use of a medical device or accessory directed to the user of the medical device“
“General and technical information provided by the manufacturer to inform the user of the medical device or IVD medical device’s intended purpose and proper use and of any contraindications, warnings, or precautions to be taken. It is provided by the manufacturer to support and assist the device users in its safe and appropriate use.”
Mit Gebrauchsanweisung ist also in der Regel ein gedrucktes Begleitmaterial gemeint, welches den Nutzer über Zweckbestimmung, Sicherheitsinformationen und die korrekte Verwendung informieren soll.
Als Synonyme werden häufig folgende Begriffe verwendet:
- Bedienungsanleitung
- Handbuch
- Operation Manual
- User Guide
- IFU (instructions for use)
Gemeint ist jeweils das gleiche.
2. Regulatorische Anforderungen an Gebrauchsanweisungen
Zahlreiche Gesetze, Verordnungen, Richtlinien, Normen und andere Vorschriften stellen Anforderungen an die Gebrauchsanweisungen. Dazu zählen:
|
Regularien |
Beschreibung |
|
Anforderungen an Gebrauchsanweisungen werden insbesondere in Anhang I Absatz 23 gestellt. | |
|
Anforderungen an Gebrauchsanweisungen werden insbesondere in Anhang I Absatz 20 gestellt. | |
| Die VO 2021/2226 regelt, unter welchen Voraussetzungen die Gebrauchsanweisung elektronisch bereitgestellt werden darf (eIFU) und welche Anforderungen damit verknüpft sind. Aktuell wird die Verordnung überarbeitet, um eIFUs auch für Laienanwender von Softwareprodukten zu ermöglichen (siehe hier). | |
|
Die Gebrauchsanweisung zählt zum User Interface. Entsprechend muss sie gemäß einem Usability-Engineering-Prozess nach der 62366-1 entwickelt werden. | |
|
Kapitel 7.9 legt fest, welche Inhalte Gebrauchsanweisungen von medizinisch-elektrischen Geräten enthalten müssen. | |
|
Die ISO 20417 ersetzt die EN 1041 und stellt insbesondere in Kapitel 6.6 Anforderungen an die Gebrauchsanweisung. | |
| IEC 81001-5-1 | Anhang E spezifiziert, welche Inhalte dem Anwender in Hinblick auf Cybersecurity mitgeteilt werden müssen. |
ISO 18113-(1-5): IVD − Bereitstellung von Informationen durch den Hersteller – Teil 1: Begriffe und allgemeine Anforderungen – Teil 2: Reagenzien für den Gebrauch durch Fachpersonal – Teil 3: Geräte für in-vitro-diagnostische Untersuchungen zum Gebrauch durch Fachpersonal – Teil 4: Reagenzien für in-vitro-diagnostische Untersuchungen zur Eigenanwendung – Teil 5: Geräte für in-vitro-diagnostische Untersuchungen zur Eigenanwendung | Die Teile 1-5 der ISO 18113 legen fest, welche Anforderungen Gebrauchsanweisungen für IVDs erfüllen müssen. |
|
IMDRF/GRRP WG/N52 FINAL:2019: Principles of Labelling for Medical Devices and IVD Medical Devices | Das Ziel dieses Dokuments der IMDRF/GRRP ist es, international harmonisierte Anforderungen an das Labelling (inklusive Gebrauchsanweisungen) aufzustellen. |
Die FDA stellt eine Reihe von Anforderungen an die Gebrauchsanweisung; im 21 CFR part 801 für Medizinprodukte und im part 809 für IVDs. | |
|
Medizinproduktehersteller sollten sich an diesen Leitfaden für Zulassungen in den USA halten. Darüber hinaus bietet er auch für Zulassungen außerhalb der USA hilfreiche Tipps für das Erstellen von Gebrauchsanweisungen. | |
|
FDA Guidance: Applying Human Factors and Usability Engineering to Medical Devices |
Analog zur 62366-1 sollten sich Hersteller bei der Entwicklung von Gebrauchsanweisung für den US-Markt an die Vorgaben dieses Leitfadens halten. |
| MDCG 2019-16 | Kapitel 4 befasst sich mit Anforderungen an die Gebrauchsanweisung hinsichtlich Cybersecurity. |
| FDA Cybersecurity Guidance Documents | Der Cybersecurity und der zugehörigen Gebrauchsanweisung hat die FDA gleich zwei Guidance-Dokumente gewidmet. |
a) Best Practice Guides zum Erstellen von Gebrauchsanweisungen
Weitere Normen und Leitfäden geben Hinweise, wie man gute Gebrauchsanweisungen schreibt:
- IEC/IEEE 82079-1: Ebenso umfangreich wie handlungsleitend ist die IEC/IEEE 82079-1 (Nachfolger der DIN EN 62079). Obwohl diese Norm keinen Branchenfokus hat, gilt sie auch für Medizinprodukte als Stand der Technik für die Erstellung von Gebrauchsanweisungen.
- AAMI TIR 49: Im Gegensatz zur 82079-1 legt die AAMI TIR 49 den Fokus auf Medizinprodukte und Hinweise zum „Design of training and instructional materials for medical devices used in non-clinical environments“.
- ANSI Z535: Die ANSI Z535 legt fest, wie sicherheitsbezogene Hinweise zu gestalten sind, beispielsweise wie Symbole und Farben zu verwenden sind.
- Die WHO Guidance: Designing instructions for use for in vitro diagnostic medical devices gibt auch für Geräte, die keine IVD sind, wertvolle Tipps zum Erstellen von Gebrauchsanweisungen. Sie gibt sogar eine Struktur vor und enthält Textvorlagen.
- Der Medical Device and Health IT Joint Security Plan version 2 (JSP2), welcher auch von der FDA referenziert wird, liefert in Anhang D eine gute Grundstruktur zum Erstellen der Cybersecurity-Dokumentation.
Mehr zu den Empfehlungen dieser Leitfäden finden Sie in den Kapiteln 4 und 5.
b) Wann es einer Gebrauchsanweisung bedarf und wann nicht
Bestimmungen der MDR
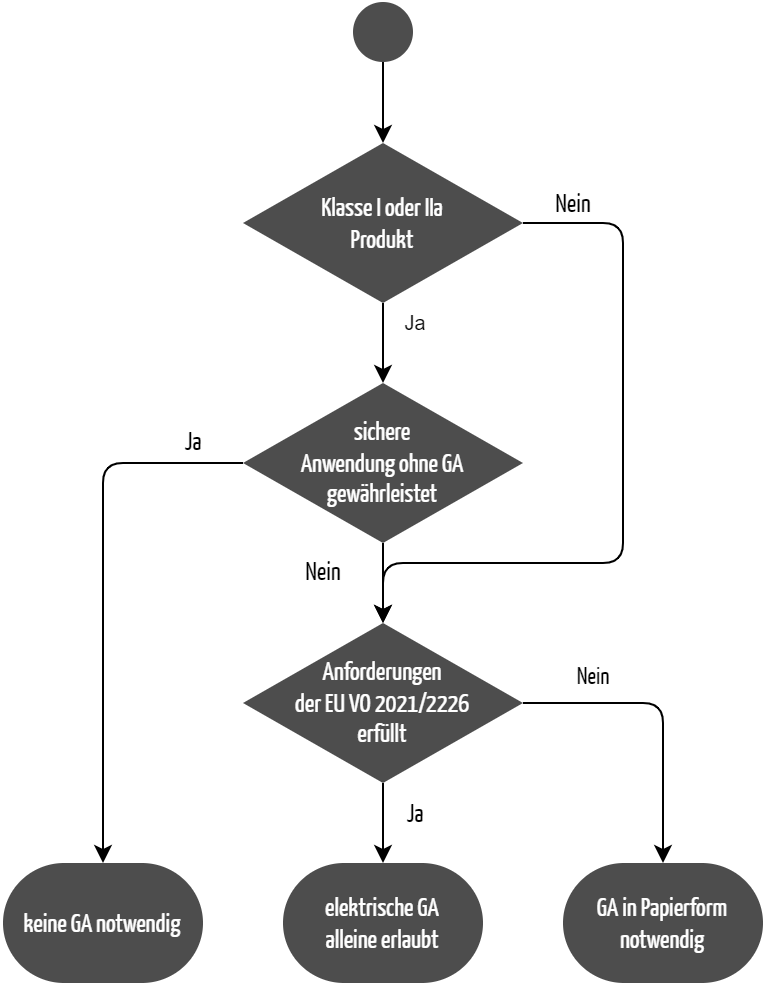
Die Medizinprodukteverordnung (MDR) erlaubt es, unter bestimmten Voraussetzungen auf die Gebrauchsanweisung zu verzichten (siehe Abbildung 1). Sie schreibt im Anhang I, Kapitel 23.1 d.:
„Die Gebrauchsanweisung wird zusammen mit dem Produkt bereitgestellt. Eine Gebrauchsanweisung ist für Produkte der Klassen I und IIa ausnahmsweise entbehrlich, wenn eine sichere Anwendung dieser Produkte ohne Gebrauchsanweisung gewährleistet ist und sofern an anderer Stelle dieses Abschnitts nichts anderes angegeben ist.“
Der Nachweis, dass das Produkt auch ohne die Gebrauchsanweisung sicher verwendet werden kann, kann auf zwei Wegen erbracht werden:
- Die Risikoanalyse des Herstellers nach ISO 14971 zeigt, dass durch eine Fehlanwendung des Produkts keine Gefährdungssituation für Anwender oder Patienten entsteht. Damit ist eine sichere Anwendung des Produkts auch ohne Gebrauchsanweisung impliziert. Dieser Nachweis birgt allerdings die Gefahr, dass man mögliche Benutzungsfehler der Anwender in der Risikoanalyse nicht berücksichtigt. Häufig beobachten Hersteller erst im Usability-Test Benutzungsfehler, an die sie vorher nicht gedacht haben.
- Usability-Tests stellen daher eine aussagekräftigere Methode zum Nachweis dar. Wenn in Usability-Tests den vorgesehenen Anwendern in der vorgesehenen Gebrauchsumgebung auch ohne Gebrauchsanweisung keine Benutzungsfehler unterlaufen, liefert das den objektiven Nachweis der sicheren Anwendbarkeit.
Bestimmungen der IVDR
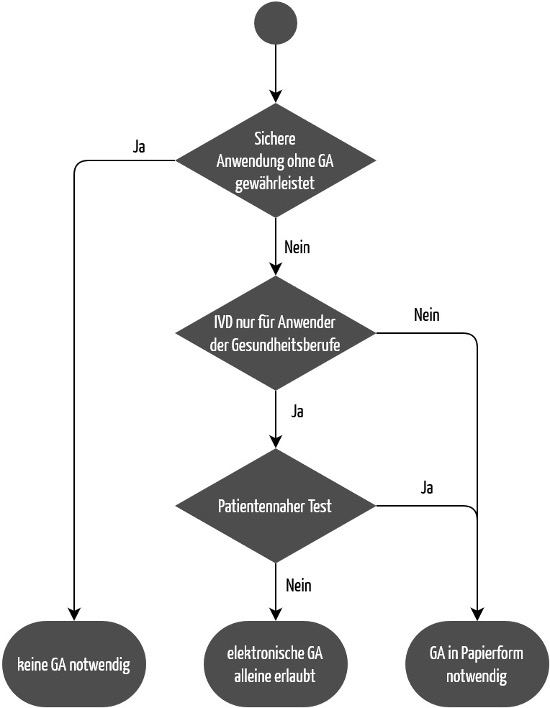
Auch bei IVDs kann unter bestimmten Voraussetzungen auf eine Gebrauchsanweisung verzichtet werden (siehe Abbildung 2). Im Gegensatz zur MDR macht die IVDR diese Ausnahme aber nicht von der Klasse des Produkts abhängig. Die IVDR schreibt in Anhang I, Kapitel 20.1.d:
„Die Gebrauchsanweisung wird zusammen mit dem Produkt bereitgestellt. In hinlänglich begründeten Ausnahmefällen sind allerdings keine Gebrauchsanweisungen erforderlich oder können diese gekürzt werden, wenn das Produkt ohne solche Anleitungen sicher und wie vom Hersteller vorgesehen verwendet werden kann.“
c) In welcher Form und welcher Anzahl müssen Gebrauchsanweisungen bereitgestellt werden?
Grundsätzlich muss die Gebrauchsanweisung in Papierform vorliegen. Ausnahmen sind unter gewissen Voraussetzungen möglich (siehe Kapitel 5d).
MDR und IVDR fordern, dass die Gebrauchsanweisung zusammen mit dem Produkt bereitgestellt wird.
Liefert der Hersteller mehrere Produkte an einen Anwender und/oder Ort aus, reicht unter folgenden Bedingungen die Bereitstellung nur einer Gebrauchsanweisung:
- Die Bereitstellung nur einer Gebrauchsanweisung wurde vorher mit dem Käufer vereinbart und
- es können jederzeit auf Nachfrage kostenlose Exemplare nachgeliefert werden.
Unter der IVDR ist die Bereitstellung nur einer Gebrauchsanweisung unter den gleichen Bedingungen möglich. Voraussetzung ist, dass es sich nicht um Produkte zur Eigenanwendung oder für patientennahe Tests handelt.
Eine Gebrauchsanweisung muss nicht als ein gebundenes Dokument vorliegen. Es kann sogar empfehlenswert sein, die Gebrauchsanweisung auf mehrere Dokumente aufzuteilen, z. B. in eine Version speziell für Laien und eine für professionelle Anwender oder Servicekräfte.
d) Bereitstellung als elektronische Gebrauchsanweisung (eIFU)
Unter der MDR hat der Hersteller die Möglichkeit, die Gebrauchsanweisung nur elektronisch zur Verfügung zu stellen. Die Verordnung 2021/2226 zu elektronischen Gebrauchsanweisungen legt fest, wann eine Gebrauchsanweisung in elektronischer Form anstatt in Papierform erlaubt ist.
Lesen Sie hier mehr zum Thema elektronische Gebrauchsanweisungen und Verordnung 2021/2226.
Die EU-Verordnung 2021/2226 zu elektronischen Gebrauchsanweisungen gilt nicht für IVDs. Stattdessen regelt die IVDR in Anhang I, Kapitel III, 20.1.f, wann Hersteller von der Papierform abweichen dürfen:
„Wenn das Produkt lediglich für den beruflichen Gebrauch bestimmt ist, kann die Gebrauchsanweisung dem Anwender in anderer Form als in Papierform (z. B. elektronisch) bereitgestellt werden, es sein denn, das Produkt ist für patientennahe Tests vorgesehen.“
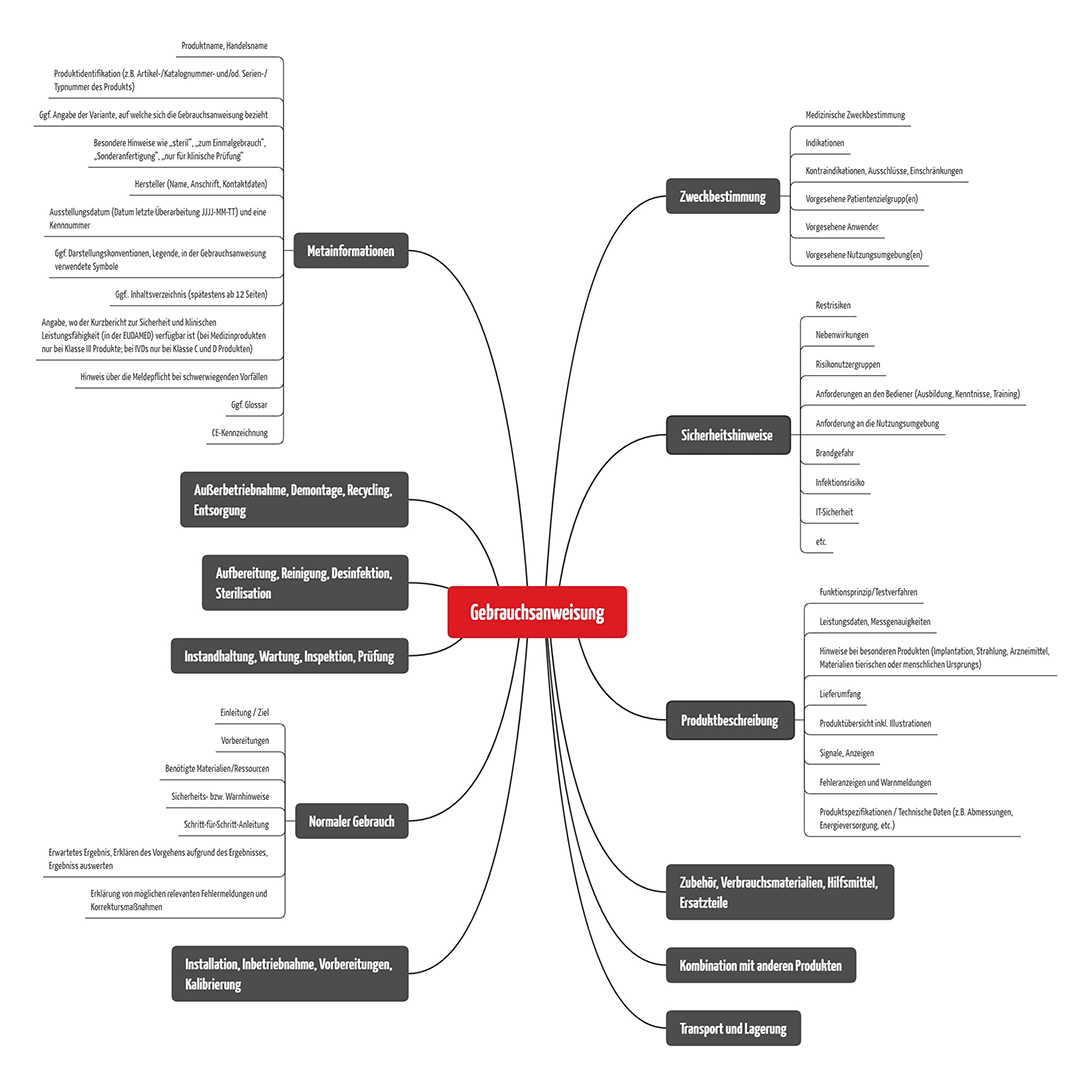
e) Inhalte von Gebrauchsanweisungen
Trägt man die Anforderung der oben genannten Regularien zusammen, ergeben sich unter anderem folgende übergeordnete Inhalte:
f) Anforderungen an die Sprache von Gebrauchsanweisungen
In Artikel 10 (11) der MDR bzw. Artikel 10 (10) der IVDR wird gefordert, dass die Gebrauchsanweisung in einer oder mehreren der vom Zielland festgelegten Sprache(n) vorliegen muss.
Belgien fordert beispielweise, dass die Gebrauchsanweisung auf Deutsch UND Niederländisch UND Französisch vorliegen muss – oder aber auf Englisch, wenn es sich um professionelle Anwender handelt und diese eine Sprache ihrer Wahl kostenfrei nachfordern können.
Im Januar 2024 hat die Europäische Kommission endlich – 7 Jahre nach der Veröffentlichung der MDR – eine Übersicht aller nationalen Sprachanforderungen in der EU herausgegeben. Diese ist sehr hilfreich und in vielen Fällen auch direkt mit dem entsprechenden Gesetz verlinkt. Auch das Graphical User Interface wird explizit genannt. Leider scheint die Herausgeber während des Schreibens ein wenig die Motivation verlassen zu haben, die Original-Gesetze zu durchsuchen (Ungarn), sie zu übersetzen und den Inhalt auf Korrektheit zu prüfen (Litauen) oder die Tabelle fertig auszufüllen (Spanien).
Nehmen Sie Kontakt zu unserem Beratungsteam auf, um eine aktuelle Liste mit Sprachanforderungen an die Gebrauchsanweisung und Kennzeichnung für Medizinprodukte und IVD innerhalb der EU zu erhalten. Diese können Sie ggf. als Ergänzung zur Liste der Europäischen Kommission nutzen.
In Deutschland regelt das Medizinproduktedurchführungsgesetz die Sprachfrage:
8 (2) „Produkte dürfen im Geltungsbereich dieses Gesetzes nur dann an Anwender und Patienten abgegeben werden, wenn die für Anwender und Patienten bestimmten Informationen in deutscher Sprache zur Verfügung gestellt werden. In begründeten Fällen dürfen die Informationen auch in englischer Sprache oder einer anderen für den Anwender des Medizinproduktes leicht verständlichen Sprache zur Verfügung gestellt werden, wenn diese Informationen ausschließlich für professionelle Anwender bestimmt sind und die sicherheitsbezogenen Informationen auch in deutscher Sprache oder in der Sprache des Anwenders zur Verfügung gestellt werden.“
In der Schweiz muss die Gebrauchsanweisung grundsätzlich auf Deutsch UND Französisch UND Italienisch vorliegen, unabhängig vom Sprachgebiet. Weniger als drei Amtssprachen oder Englisch ist für Fachpersonen erlaubt, sofern diese damit einverstanden sind und sich daraus keine Risiken ergeben (siehe MepV Art. 16).
g) Anforderungen an die Übersetzung der Gebrauchsanweisungen
Die MDR bzw. IVDR stellen keine expliziten Anforderungen an die Übersetzung der Gebrauchsanweisung. Die Gebrauchsanweisung muss jedoch für die vorgesehen Nutzer in allen Zielsprachen verständlich sein. Im Anhang I Absatz 23.1 a der MDR heißt es dazu:
„Insbesondere ist die Gebrauchsanweisung so zu verfassen, dass sie von dem vorgesehenen Anwender ohne Schwierigkeiten verstanden wird […]“.
Das stellt besondere Anforderungen an die Übersetzung der Gebrauchsanweisung von der Originalsprache in die Zielsprache.
Anforderungen an den Übersetzungsprozess sowie den Linguisten kommen unter anderem von der IEC/IEEE 82079‑1 sowie vom Qualitätsstandard für Übersetzungsdienstleister ISO 17100:2015. Der Linguist sollte beispielweise folgende Kompetenzen besitzen:
- Er sollte theoretisches Wissen und praktische Erfahrung beim Übersetzen von medizinischen Texten besitzen.
- Er sollte die Originalsprache fließend beherrschen.
- Er sollte Muttersprachler der Zielsprache sein.
- Er sollte über eine gewisse Mindesterfahrung im Bereich der angeforderten Übersetzung verfügen und muss mit der produktspezifischen Terminologie vertraut sein.
Insbesondere die Anforderung, Muttersprachler der Zielsprache zu sein, dürfte bei 24 Amtssprachen in der EU von Herstellern selbst nur schwer zu erfüllen sein. Hierbei empfiehlt es sich, Übersetzungsdienstleistern zu vertrauen.
Stellt der Übersetzungsdienstleister für den Medizinproduktehersteller einen kritischen Lieferanten dar, empfehlen wir bei der Auswahl des Dienstleisters auf folgende Kriterien zu achten. Der Übersetzer sollte Erfahrung im Bereich der Medizintechnik vorweisen können und auf den Bereich Gebrauchsanweisung im Rahmen der MDR bzw. IVDR spezialisiert sein. Er sollte nach dem Qualitätsstandard für Übersetzungsdienstleister ISO 17100 zertifiziert sein. Zudem sollte er nach einem Quality-Management-System arbeiten, bspw. angelehnt an ISO 9001 oder ISO 13485.
3. Entwicklungsprozess der Gebrauchsanweisung
Die IEC 62366-1 und der FDA-Leitfaden „Human Factors Engineering“ fordern einen Prozess für die Entwicklung eines User Interfaces (UI). Die Gebrauchsanweisung ist integraler Bestandteil des User Interface. Hersteller müssen deswegen den Usability-Engineering-Prozess auch auf die Gebrauchsanweisung anwenden, was Usability-Tests der Gebrauchsanweisung mit repräsentativen Nutzern einschließt.
Erfahren Sie mehr zum Usability-Engineering-Prozess nach 62366-1 in unseren Blogartikeln IEC 62366-1:2015 Neues zur Usability-Norm und Summative Evaluierung: Auf diese Punkte müssen Sie achten.
Zusätzlich geben sowohl die IEC 82079-1 als auch die AAMI TIR 49 spezifische Empfehlungen für die Etablierung eines Prozesses zur Erstellung von Gebrauchsanweisungen.
Der Prozess für die Entwicklung von Gebrauchsanweisungen sollte unter anderem folgende Schritte beinhalten:
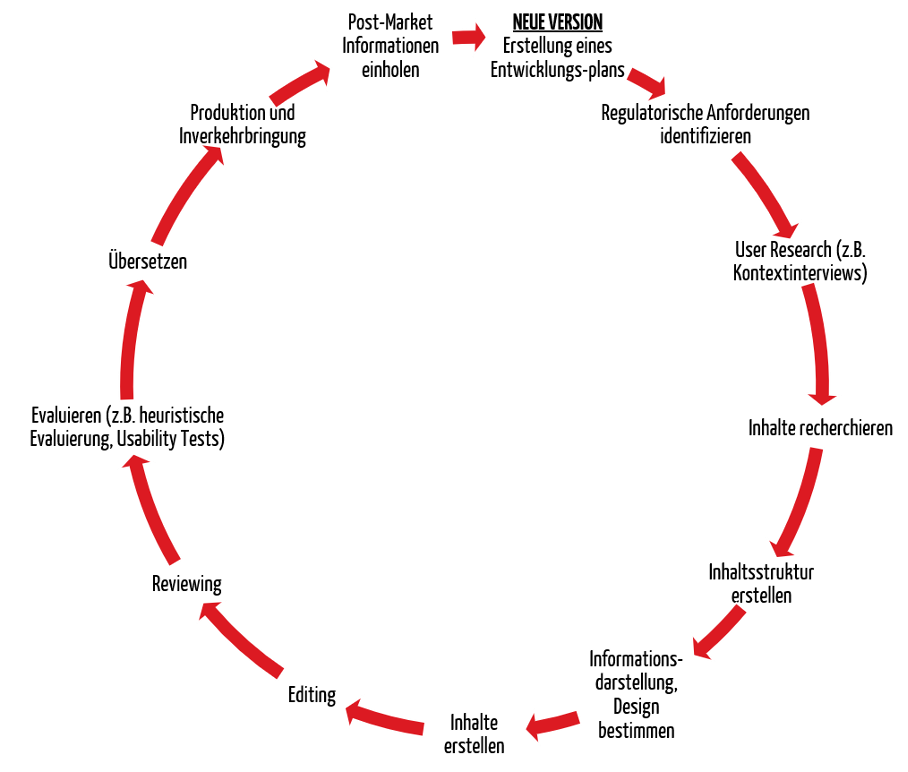
Der Prozess sollte iterativ angewendet werden. Das heißt, dass zu wichtigen Meilensteinen eine Evaluierung stattfinden und die Ergebnisse der Evaluierung als Input für die Weiterentwicklung dienen sollten.
4. Typische Fehler und ihre Folgen
a) Auf welche Fehler die Berater und Auditoren des Johner Instituts regelmäßig stoßen
- Die Gebrauchsanweisung ist inhaltlich falsch.
- Sie stimmt nicht (mehr) mit dem Produkt überein (das ist oft bei Software-Updates der Fall).
- Es ist unklar, auf welches Produkt bzw. auf welche Variante sich die Gebrauchsanweisung bezieht und welche Version der Gebrauchsanweisung vorliegt.
- Begriffe sind nicht definiert, eine konsistente Terminologie fehlt, was Missverständnisse fördert.
- Die Texte sind unverständlich, Sätze sind zu lang, viele Formulierungen sind im Passiv und zu abstrakt. Die Sprache verwendet technische Begriffe und orientiert sich nicht am Vokabular der Zielgruppe.
- Die Struktur fehlt und ist nicht nachvollziehbar, was Navigation und Orientierung erschwert.
- Grafiken oder andere Formen der Visualisierung fehlen.
- Schriftgrößen sind zu klein und Farbkodierungen fehlen bzw. werden inkonsistent oder gar normenwidrig verwendet. Das gleiche gilt für Textgrößen und Textauszeichnungen sowie für die Seitenformatierung und die Verwendung von Elementen wie Symbolen.
- Die Übersetzungen fehlen oder sind fehlerhaft. Google Translate ist ein No-Go.
- Für verschiedene Produktvarianten und Kombinationen mit anderen Produkten gibt es nur eine einzige Gebrauchsanweisung, die entsprechend schwer zu verstehen ist.
- Inhalte, die für das Verständnis und die sichere Anwendung notwendig sind, fehlen. Es werden insbesondere nicht alle Aufgaben sowie nicht der komplette Lebenszyklus von der Installation bis zur Außerbetriebnahme und Entsorgung beachtet.
- Überflüssige Informationen, z. B. über den Hersteller, blähen die Gebrauchsanweisung auf.
- Die Gebrauchsanweisung wurde nicht von qualifiziertem Personal erstellt, sondern z. B. von ungeübten Entwicklern.
- Die Hersteller überprüfen die Wirksamkeit der Gebrauchsanweisung nicht, z. B. im Rahmen eines Usability-Tests.
- Die Zielgruppe wird nicht ausreichend berücksichtigt.
- Die Hersteller haben keinen Prozess festgelegt, wie dem Nutzer auch nach Auslieferung Fehler in der Gebrauchsanweisung kommuniziert werden.
b) Welche Konsequenzen mangelhafte Gebrauchsanweisungen haben können
In Dutzenden Usability-Tests haben unsere Berater immer wieder Benutzungsfehler aufgrund von mangelhaften Gebrauchsanweisungen beobachtet. Probleme in Usability-Tests aufgrund mangelhafter Gebrauchsanweisungen können die Produktzulassung verzögern.
Sollte Ihr Produkt mit einer mangelhaften Gebrauchsanweisung bereits in Verkehr gebracht worden sein, können im schlimmsten Fall Schäden für Patienten, Anwender oder Dritte die Folge sein.
5. Fünf Tipps zum Schreiben von Gebrauchsanweisungen
1. Tipp: Übertragen Sie diese Aufgabe an Profis
Überlassen Sie das Schreiben von Gebrauchsanweisungen nicht Entwicklern oder Produktmanagern, die nicht dafür qualifiziert sind. Sie benötigen darauf spezialisierte Autoren, Redakteure, Übersetzer, Lektoren, Illustratoren und Prüfer.
2. Tipp: Spezifizieren Sie Ihre Leser und Ihre Ziele
Eine Gebrauchsanweisung ist nicht per se gut oder schlecht. Die Gebrauchsanweisung muss für die in der Zweckbestimmung spezifizierten Anwender geschrieben sein. Eine Gebrauchsanweisung für das medizinische Fachpersonal unterscheidet sich von der für einen Laien genauso wie von der für einen Servicetechniker.
Sie sollten Ihre Ziele, die Sie mit der Gebrauchsanweisung erreichen wollen, schriftlich formulieren. Welche Nutzungsfehler und damit welche Risiken möchten Sie minimieren? Bei welchen Aufgaben soll eine sichere Nutzung gewährleistet werden?
3. Tipp: Nutzen Sie eine konsistente Struktur und einen Redaktionsleitfaden
Eine konsistente Darstellung erleichtert das Lesen. Ein Redaktionsleitfaden sollte regeln, dass eine Anleitung die folgenden Elemente beinhaltet:
- Thema/Überschrift
- Beschreibung
- Ziel der Handlung
- Voraussetzung
- Zustand
- Handlung
- Zu erzielendes Ergebnis
- Warnung
- Beispiel
- Bilder und Tabellen jeweils mit zugehörigen Beschriftungen
Beim Gestalten von Gebrauchsanweisungen sollten Sie u. a. folgende Punkte berücksichtigen:
- Nummerieren Sie die einzelnen Schritte in arabischer Zahlenschrift.
- Verfassen Sie Anweisungen im Imperativ, in der Aktivform und in einfacher Sprache.
- Verwenden Sie Verben und keine Substantivierungen.
- Beschreiben Sie pro Schritt nur eine Handlung (bzw. maximal drei logisch und eng verknüpfte Handlungen). Geben Sie pro Satz nur eine Anweisung.
- Wenn Sie Illustrationen einfügen, sollten diese dem erläuternden Text eindeutig zuzuordnen sein.
Regelmäßig formulieren Hersteller zu wenige handlungsleitende Anweisungen.

4. Tipp: Verstehen Sie das Schreiben als einen (kontinuierlichen) Prozess
Das Schreiben von Gebrauchsanweisungen ist ein Teamsport, der viele Arbeitsschritte durchläuft, die definiert und geplant sein sollten.
Wie das Produkt, so muss auch eine Gebrauchsanweisung kontinuierlich weiterentwickelt werden.
5. Tipp: Beachten Sie die ISO 14971, die IEC 62366-1 bzw. den FDA-Leitfaden ‚Human Factors Engineering‘
Die Gebrauchsanweisung ist Teil des Produkts. Damit unterliegt sie den regulatorischen Anforderungen an die Gebrauchstauglichkeit. Die IEC 62366:2006 enthielt für die Begleitmaterialien sogar ein eigenes Kapitel.
Die IEC 62366-1:2015 verlangt, diese Materialien genauso wie das Produkt selbst zu behandeln. Dazu zählt auch die Überprüfung der Gebrauchstauglichkeit und der Wirksamkeit von Maßnahmen – hier in Form von Hinweisen in der Gebrauchsanweisung.
Professionell arbeitende Firmen kombinieren dazu mehrere Methoden:
- Usability-Tests / teilnehmende Beobachtungen
- Befragungen
- Selbsteinschätzungen
- Gutachten, Inspektionen, Überprüfung durch Experten
- Analyse von Rückmeldungen zu Vorgängerversionen der Gebrauchsanweisung
Das Johner Institut prüft die Gebrauchsanweisungen oft im Rahmen der formativen und summativen Evaluation der Gebrauchstauglichkeit.
6. Tipp: Cybersecurity-Dokumentation als risikominimierende Maßnahme
Gemäß des Defense-In-Depth Konzepts ist es unerlässlich, die Anwender Ihrer vernetzten Produkte ausreichend über die Cybersecurity aufzuklären und dadurch mögliche Risiken so weit wie möglich zu reduzieren. Gerade die FDA fordert hier ein hohes Maß an Transparenz, welches durch eine ausführliche Gebrauchsanweisung erreicht werden kann. Im Detail sind dies:
- Anweisungen und Spezifikationen (z. B. Malware-Schutz)
- Diagramme
- Eine Liste der Netzwerk-Ports und Schnittstellen
- Anforderungen an die Infrastruktur
- Die SBOM
- Ein möglicher Download-Prozess
- Verhalten des Produktes bei Security-Vorfällen
- Beschreibung der Sicherheitsfunktionen
- Backup und Wiederherstellung
- Beschreibung der sicheren Konfiguration nach Versenden (Passwörter ändern, Firewall Einstellungen etc.)
- Logging Mechanismus
- End of Support / End of Life
- Sichere Entsorgung (zum Beispiel Bereinigung von sensiblen Daten)
Hier verweist sie explizit auf den Medical Device and Health IT Joint Security Plan (JSP2), welchen Hersteller als Grundlage nutzen können, um diese Informationen zu übermitteln. Anhang D kann Ihnen hier als gutes Beispiel dienen, welches Sie in Übereinstimmung mit Ihrer Risikoanalyse spezifisch auf Ihr Produkt anpassen können – dieses Vorgehen empfiehlt sich auch für Europa, da sich die Vorgaben mit der IEC 81001-5-1 großenteils decken.
Auch das MDS2-Formular gewinnt international immer mehr an Bedeutung. Viele Anwender setzen dies mittlerweile als Kaufbedingung voraus.
6. Fazit
Eine Gebrauchsanweisung zu schreiben erfordert das gleiche Maß an Professionalität wie das Entwickeln des (eigentlichen) Produkts. Diese Aufgabe darf nicht als notwendiges Übel am Ende einer Entwicklung „schnell“ noch an Personen übertragen werden, die dafür nicht ausreichend qualifiziert sind.
Die MDR, IVDR und Normen stellen zwar Anforderungen an die Informationen, die eine Gebrauchsanweisung enthalten muss; konkrete Hilfestellungen, wie gebrauchstaugliche Gebrauchsanweisung zu schreiben sind, finden sich hingegen in anderen Normen wie der IEC 82097-1 oder dem AAMI TIR 49.
Hersteller sollten unbedingt beachten, dass die Wirksamkeit der Gebrauchsanweisung ebenso geprüft werden muss wie andere risikominimierende Maßnahmen, die insbesondere die Gebrauchstauglichkeit betreffen. Daher sollten Gebrauchsanweisungen im Rahmen der formativen und summativen Bewertung der Gebrauchstauglichkeit gemäß IEC 62366-1 ebenfalls bewertet werden.
Die Expertinnen und Experten des Johner Instituts in Frankfurt und Washington D.C. helfen Ihnen mit Beratung und Templates beim Verfassen und beim Prüfen auch englischsprachiger Gebrauchsanweisungen. Kontaktieren Sie uns.
Versionshistorie:
- 2024-08-23: Ergänzung weiterer Normen und Guidance Documents, Anpassung Kapitel 2.f), Ergänzung eines weiteren Tipps in Kapitel 5, redaktionelle Änderungen
- 2022-02-18: Komplette Überarbeitung



Der Link zur „AAMI TIR 49“ ist fehlerhaft
Danke für den Tipp, lieber Herr Fiege! Da hatte sich eine Klammer in die URL eingeschmuggelt. Dank Ihres Hinweises konnten wir das gleich „fixen“.
Guten Abend.
Muss eine Gebrauchsanweisung aktualisiert werden, wenn das Produkt sich nicht geändert hat aber die gesetzlichen Verordnungen?
Beispiel: In der GA von 2015 steht: „Das Produkt ist nicht einweisungspflichtig.“ Mit dem Inkrafttreten der neuen MPBetreibV in 1/17 besteht die Verpflichtung in (fast) alle Medizinprodukte einzuweisen.
Muss der Hersteller seine GA an die neue Gesetzeslage anpassen? Wo ist die Quelle hierfür?
Danke vorab.
Dr. Thomas Castner
Sehr geehrter Herr Dr. Castner,
wenn Sie Produkte in den Verkehr bringen, müssen diese die regulatorischen Anforderungen erfüllen. Das steht im Gesetz direkt, indirekt auch in der MDD. Mit Ihrer Konformitätserklärung bestätigen Sie das auch.
Das MPG schreibt u.a. „Über die Beschaffenheitsanforderungen hinausgehende Bestimmungen, die das Betreiben oder das Anwenden von Medizinprodukten betreffen, bleiben unberührt.“ Darüberhinaus verpflichtet auch die ISO 13485 die Hersteller regulatorische Anforderungen zu erfüllen.
In der Hoffnung, etwas geholfen zu haben, und mit den besten Grüßen
Christian Johner
Wir liefern zu jedem Medizinprodukt 2 Bedienungsanleitungen in gedruckter Form aus. Auch wenn gleiche Produkttypen ausgeliefert werden. So kann es sein, dass Kunden bei 20 gleichen Geräten 40 Bedienungsanleitungen bekommt. Es handelt sich hier um Klasse IIa Geräte. Lt. MPG sollen 2 Bedienungsanleitungen pro Gerätetyp geliefert werden.
Nun meine Frage:
a.) Können flex. Endoskope mit elektronischen Bedienungsanleitungen geliefert werden?
b.) Reicht es, pro Produkttyp max. 2 Bedienungsanleitungen zu liefern? (Reduktion von Ressourcen und Müllvermeidung) Zum großen Teil werden überflüssige Anleitungen vor Ort vom Techniker entsorgt.
Gruß,
Stephan Kohl
Lieber Herr Kohl,
danke für Ihre Fragen zu den elektronischen Gebrauchsanweisung. Ich habe dazu einen Beitrag zu den elektronischen Gebrauchsanweisungen verfasst. Könnten Sie einen Blick darauf werfen und mich wissen lassen, ob das die erste Frage beantwortet?
Eine gesetzliche Anforderung, die die Anzahl der Gebrauchsanleitungen regelt, insbesondere eine, die mehr als eine verlangt, ist mir nicht bekannt.
Bitte haken Sie bei Rückfragen gerne nach.
Viele Grüße, Christian Johner
Guten Morgen Herr Johner.
Wenn man die Gruppe der Medizinprodukte in der VO 207/2012 und dementsprechend in Ihrem Beitrag genau betrachtet, so werden MP der Klasse I ausgeschlossen. Aus meiner Sicht ist dies jedoch nicht nachvollziehbar, da MP mit einer potentiell höheren Gefährdungsklasse eingeschlossen werden.
Wie stehen Sie zu diesem Sachverhalt?
Zu Herrn Kohl: Eine Anforderung für mehr als eine Gebrauchsanweisung pro Produkt ist mir ebenfalls völlig neu.
Für Ihre Unterstützung im Voraus besten Dank.
Viele Grüsse
Thomas Castner
Sehr geehrer Hr. Johner,
danke für Ihre Antwort.
Da haben Sie natürlich Recht. Da hat sich wohl in unserem System eine kapitale Fehleinschätzung eingeschlichen.
Gruß,
Stephan Kohl
Sehr geehrter Herr Johner,
ist es aus regulatorischer Sicht erlaubt eine ausführliche Gebrauchsanweisung auf deutsch und englisch zur Verfügung zu stellen und gleichzeitig in andere EU Länder einer verkürzte Anleitung zu liefern ( natürlich mit dem in der Norm geforderten Inhalt ).
Sehr geehrter Herr Fecker,
wenn die kurze Gebrauchsanweisung die regulatorischen Forderungen (insbesondere mit Bezug zum Risikomanagement) erfüllt, spricht nichts dagegen, zusätzlich eine noch ausführlichere GA zur Verfügung zu stellen.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
auch die Schweiz erlaubt eine englischsprachige Bedienungsanleitung für professionelle Benutzer (Fachpersonal).
Freundliche Grüsse aus der Schweiz
Daniel Schenk
Das ist ein sehr wertvoller Hinweis, lieber Herr Schenk!
Ich aktualisiere die Webseite sofort.
Vielen Dank! Beste Grüße aus Kreuzlingen-Nord (KN), Christian Johner
Sehr geehrter Herr Johner,
haben Sie einen Tipp für unser junges Startup, wo wir die Mindestanforderungen für Gebrauchsanweisungen für die jeweiligen EU-Staaten oder eine Art Leitfaden finden können?
Vielen Dank und mit freundlichen Grüßen
Felix Ziegler
In der EU sind die Anforderungen EU-weit geregelt.
Die MDR benennt diese z.B. im Anhang I Kapitel 23.
Auf der Webseite habe ich weitere regulatorisch relevante Dokumente genannt, beispielsweise zu den Sprachen.
Der wichtigste Hinweis ist, dass die Inhalt der Gebrauchsanweisung mit den Ergebnissen des Risikomanagements abgestimmt sein müssen.
Geben Sie einfach Bescheid, wenn wir für Sie noch weiter durch-deklinieren können.
Sehr geehrter Herr Johner,
muss eine in England konforme und zertifizierte GA für ein Medizinprodukt für den deutschen Markt original-übersetzt werden oder reicht es aus, die genannten Kriterien für eine GA allgemein in deutscher Sprache zu erfüllen.
Vielen dank und mit freundlichen Grüßen
Julius Fach
Sehr geehrter Herr Fach,
danke für Ihre Nachricht! Ich bin noch nicht ganz sicher, ob ich alles verstehe. Eine Zertifizierung von GAs ist mir nicht bekannt — und auch nicht notwendig. Sie erklären die Konformität des Produkts und damit der GA. Wenn diese GA die gesetzlichen Anforderungen erfüllt (und die anderen Voraussetzungen auch erfüllt sind), dürfen Sie das Produkt auch im deutschsprachigen Raum in den Markt bringen.
Möglicherweise ist meine Antwort noch unvollständig. Haken Sie dann einfach nach.
Viele Grüße, Christian Johner
Sehr geehrter Herr Dr. Johner,
Sie verweisen in Ihrem Artikel auf die erforderlichen Sprachen für eine GA nach „MDEG – 2008-12 – II-6.3. Mandatory Languages Requirements for Medical Devices update Sept.08“. Ist beim Übergang auf die MDR mit einer Änderung / einem Update der Liste zu rechnen?
Wissen Sie, ob die den NB daran gearbeitet wird?
Viele Grüße
Norbert Geerkens
Sehr geehrter Herr Geerkens,
ich vermute, dass es ein Update geben wird, mir ist aber nicht bekannt, dass die NBs daran arbeiten. Meines Erachtens wäre das auch eher eine Aufgabe der MDCG.
Solange — auch nach dem 25.05.2020 — werden wir mit den bisherigen Dokumenten weiterarbeiten, bis neue veröffentlicht wurden.
Ich würde verstärkt darauf achten, wenn es GAs für Laien gibt. Die Anforderungen an die GAs durch Anhang I Absatz 23 sind doch sehr umfangreich geworden.
In anderen Worten: Ich halte eher die Inhalte der GAs für überarbeitungswürdig als die Liste der Sprachen, in der sie übersetzt werden müssen.
Viele Grüße, Christian Johner
Guten Tag Herr Johner,
ich hätte da eine Frage bezüglich der sprachlichen Anforderungen an die GA.
Wenn ich in der EU vorerst nur in bestimmten Märkten (Z. B. D, F, GB) auf den Markt gehen will, dann genügt doch wohl die Übersetzung der GA vom Deutschen ins Französische und Englische, um dann eine CE-Kennzeichnung anbringen zu können, oder?
Will ich eine weiter Registrierung in Italien machen, dann muss ich erst die GA erneut übersetzen, um die nationale Registrierung machen zu können, richtig?
Ich will hier nur sicher gehene, dass ich nicht das gesamte Labeling bereits in allen EU Amtssprachen übersetzt haben muss, wenn ich in einem Mitgliedsland der EU mein Medizinprodukt in Verkehr bringen will?
Das wäre doch schon ein zu großer Aufwand für kleine Unternehmen.
Ich bin gespannt auf Ihre Antwort!
Mit freundlichen Grüßen
Magdalena Kedwani
Sie müssen nur die Sprachen anbieten, die für die Länder relevant sind, in denen Sie Ihre Produkte verkaufen. Es gibt keine Anforderung, dass Sie ganz zu Beginn bereits alle Sprachen zur Verfügung stellen können. Schließlich sin das unternehmerische Entscheidungen, die schwer vorhersehbar sind.
Allerdings kann eine neue Sprache als Änderung des Produkts betrachtet werden. Hier müsste man sich den Einzelfall insbesondere die Klassifizierung und das Konformitätsbewertungsverfahren ansehen.
Sehr geehrter Herr Johner,
herzlichen Dank für Ihre Antwort.
Mit freundlichen Grüßen
Magdalena Kedwani
Hallo Herr Johner,
Bestandteil in Gebrauchsanleitung ist die Erklärung verwendeter Symbole. Üblich ist am Ende oder Anfang von IfU eine Tabelle oder Liste mit den meist aus den ISO verwendeten Symbolen und einer Erklärung dazu.
Speziell bei den zu beachtenden Temperaturbereichen werden auf Produktetiketten die ISO Symoble verwendet und ergänzt um das obere und untere Temperaturlimit, soweit ein Bereich angegeben werden soll
Ist es dann erforderlich, in der Legende das Temperatursymbol nochmals inklusive Minimum- und Maximum Temperatur zu listen oder reicht dort das Temperatursymbol alleine mit dem Hinweis, dass es bei diesem Symbol um einen Hinweis auf Lager bzw Transporttemperatur handelt?
Wenn Minimum und Maximum Temperatur nochmals extra in das Temperatursymbol in die Legende eingepfelgt werden muss , wo wird dies in den ISO Standards gefordert?
Vielen Dank für Ihre Unterstützung bei Klärung dieser Frage.
Susanne Jäkel
Sehr geehrte Frau Jäckel,
danke für die Frage!
Es gibt keine explizite regulatorische Anforderungen, dass / wie die Symbole erklärt werden solle. Allerdings sind Sie verpflichtet, die Risiken auch durch mangelnde Gebrauchstauglichkeit des Produkts zu beherrschen. Die IfU wird als Teil des Produkts betrachtet.
D.h. Sie müssten analysieren, ob es Risiken dadurch gibt, dass die von Ihnen vorgesehenen Anwender in dem von Ihnen vorgesehenen Nutzungskontext die Symbole nicht kennen, die IfU nicht lesen oder die Hinweise der IfU nicht verstehen.
Darüber, ob im Streitfall ein Richter die explizite Erläuterung der Symbole als „state of the art“ einschätzt oder/und akzeptiert, dass Sie wie der EN 14971:2012 gefordert die Risiken tatsächlich so weit wie irgendmöglich reduziert haben, lässt sich nur spekulieren.
Fazit: Die Antwort ergibt sich v.a. aus dem Risikomanagement.
Beste Grüße, Christian Johner.
Guten Abend Herr Johner,
die MDR fordert in Anhang I, Abschnitt 23.4 z, dass die Gebrauchsanweisung einen Hinweis an den Anwender und/oder den Patienten enthalten muss, dass alle im Zusammenhang mit dem Produkt aufgetretenen schwerwiegenden Vorfälle dem Hersteller und der zuständigen Behörde des Mitgliedstaats, in dem der Anwender und/oder der Patient niedergelassen ist, zu melden sind.
Dazu hätte ich folgende Fragen:
1. Wenn für ein Produkt eine Gebrauchsanweisung nicht zwingend erforderlich ist, und darauf verzichtet wird, muss dann dieser Hinweis auf der Verpackung des Produkts angebracht werden?
2. Wenn einem Produkt eine grafische Gebrauchsanweisung beigelegt wird (in der grafischen Gebrauchsanweisung wird inkl. Downloadlink auf die ausführliche Textgebrauchsanweisung verwiesen), müssen beide Gebrauchsanweisungen diesen Hinweis enthalten, oder ist er auf der ausführlichen Version ausreichend?
Vielen Dank im Voraus für Ihre Hilfe.
Mit freundlichen Grüßen,
Maximilian
Sehr geehrter Maximilian,
zu Ihren Fragen:
Beste Grüße, Christian Johner
Sehr geehrte Damen und Herren,
bezugnehmend auf den BfArM Leitfaden zur Zulassung von DiGAs wird eine Gebrauchsanweisung nach Medizinprodukterecht verlangt (S. 22). Darf man gemäß Ihres Beitrags bzgl 207/2012 jeder DiGA Hersteller den Nutzern eine Gebrauchsanweisung in Papierform zukommen lassen muss? Gibt es im Zusammenhang mit DiGAs eine Ausnahme?
Sehr geehrter Herr Grahammer,
danke für Ihre Frage! Mir erschließt sich daraus nicht ganz, ob Sie fragen, ob man es darf oder muss.
Die 207/2012 erlaubt in den Ausnahmen, den der Artikel, den Sie zitieren, beschreibt, eine elektronische Gebrauchsanweisung. Diese Verordnung zählt auch zum Medizinprodukterecht. Damit dürfen Digas von dieser Möglichkeit Gebrauch machen. Es steht aber nirgends, dass man kein Papier schicken darf.
In der Hoffnung, trotzdem Ihre Frage beantwortet zu haben, und mit vielen Grüßen, Christian Johner
Guten Tag Herr Johner,
welcher Zweck steckt hinter der MDR (23.1)-Forderung, die IFU auf der Homepage des Herstellers zusätzlich zur Papierversion anzubieten?
Geht es hier um Safety-Aspekte (Hersteller ohne Homepage hätten dann kein Sicherheits-Problem)? Oder geht es um die Vereinfachung (=Kostensenkung) von IFU-Ersatzlieferungen, was dann aber keine Forderung der MDR sein sollte, sondern optional angeboten werden könnte?
Was steckt dann noch hinter dem Zwang, die IFU auf der Homepage anzubieten? Ist damit auch die Verfügbarkeit, Auffindbarkeit etc der IFU auf der Homepage über die Usabiliy zu adressieren? Oder geht es dann eher um Marketing-Material (was inhaltlich nicht falsch sein darf, aber bzgl Verfügbarkeit keine regulatorische Rolle spielt)
Mit freundlichen Grüßen,
Jörg Bigalke
Sehr geehrter Herr Bigalke,
Herr Johner hat mich gebeten, auf Ihre Frage zu antworten. Ich hoffe Sie sind mit meiner Antwort zufrieden, ansonsten haken Sie gerne nach.
Die MDR fordert in Anhang I, 23.1, dass „[…] alle für den Anwender oder gegebenenfalls dritte Personen relevanten Informationen über die Sicherheit und Leistung des Produkts“ auch auf der Webseite des Herstellers bereitzustellen sind, falls es eine Webseite gibt. Es wird hier zwar nicht explizit gefordert, dass die Gebrauchsansweisung online verfügbar sein muss, dies kann jedoch als implizit angesehen werden, da die Gebrauchsanweisung in den allermeisten Fällen relevante Informationen über die Sicherheit bzw. korrekte und sichere Anwendung des Produkts enthält.
Einen Hinweis, was die MDR mit dieser Forderung bezwecken möchte, liefert die VERORDNUNG (EU) Nr. 207/2012 über elektronische Gebrauchsanweisungen für Medizinprodukte. Dort heißt es: „Umweltbelastung kann dadurch vermieden und die Wettbewerbsfähigkeit der Medizinprodukteindustrie durch die Kostenersparnis verbessert werden, während gleichzeitig das Sicherheitsniveau beibehalten oder angehoben wird.“
Tatsächlich bietet die zusätzliche Bereitstellung von Gebrauchsanweisungen online meiner Meinung nach einige sicherheitsrelevante Vorteile:
1. Im Verlustfall kann die die Gebrauchsanweisung schnell online eingesehen werden.
2. Die Gebrauchsanweisung kann beliebig oft reproduziert werden und ist somit nicht nur einmal an einem Ort verfügbar.
3. Das Produkt ist besser identifizierbar, was dem Anwender hilft das richtige Produkt für seine Zwecke zu erwerben.
Wenn Hersteller ihre Gebrauchsanweisung online bereitstellen, müssen sie die Anforderungen von Artikel 7 b, e und g der VERORDNUNG (EU) Nr. 207/2012 erfüllen. Dieses sind:
b) die Website ist gegen unerlaubtes Eindringen durch Hard- oder Software geschützt;
e) die Website entspricht den Vorschriften der Richtlinie 95/46/EWG;
g) auf der Website finden sich alle früheren Versionen der in elektronischer Form herausgegebenen Gebrauchsanweisung sowie das jeweilige Veröffentlichungsdatum;
Mit besten Grüßen
Nils Becker
Hallo Hr. Dr. Johner,
wir beschäftigen uns derzeit mit der Interpretation der MDR Anforderung aus Annex I, 23.4. (c):
– gegebenenfalls nähere Angaben zu dem zu erwartenden klinischen Nutzen
Wie würden Sie an der Stelle „gegebenenfalls“ interpretieren? Wann wäre eine solche Angabe erforderlich bzw. wann nicht?
Herzliche Grüße
Wolfgang Decker
Großartige Frage, Herr Decker!
Solche vagen Rechtsbegriffe sind immer ein Problem.
Die Antwort findet sich in diesem Fall in der englischen Übersetzung: „where applicable“. D.h. immer, wenn es einen klinischen Nutzen gibt, dann ist der anzugeben. Das steht wahrscheinlich wegen der „Anhang XVI Produkte“ so drin.
Nochmals danke für die Frage!
Viele Grüße, Christian Johner
Sehr geehrter Herr Dr. Johner,
in der GBA ist eine Kennnummer anzugeben. Welche Kennnummer ist hier gemeint?
Sehr geehrter Herr Heider,
danke für Ihre Frage!
Die MDR nutzt den Begriff der Kennnummer in zwei Kontexten:
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
ich hätte eine Frage bezüglich der Übersetzung der Gebrauchsanweisung. Welche Normen muss der Übersetzungsdienstleister erfüllen um für die Ubersetzung der GA herangezogen zu werden?
Gibt es da auch eine Vorschrift dazu?
Vielen lieben Dank für Ihre Antwort.
Sehr geehrter Herr Cossmann,
gerne beantworte ich Ihre Frage als Experte für Gebrauchsanweisungen beim Johner Institut.
Weder die MDR noch die harmonisierten Normen stellen konkrete Anforderungen an die Übersetzung bzw. den Übersetzungsdienstleister. Die MDR verlangt lediglich, dass die Gebrauchsanweisung für die vorgesehenen Nutzer verständlich sein muss.
In der Übersetzungsindustrie greift vor allem die ISO 17100:2015, welche bestimmte Mindestvorgaben für Linguisten und Prozesse bereit hält, die Dienstleister für Übersetzungs- und mehrsprachige Inhaltslösungen einzuhalten haben. Meine Empfehlung wäre daher, sich einen Dienstleister mit entsprechendem Zertifikat zu suchen. Ein ISO 13485 Zertifikat kann weiteren Aufschluss über die Kompetenz des Dienstleister im Medizinproduktebereich liefern.
Falls Sie Hilfe bei der Suche eines entsprechenden Dienstleisters wünschen, helfe ich gerne weiter.
Herzliche Grüße
Nils Becker
Guten Tag Herr Johner,
ich habe eine Frage zur VO 207/2012, Artikel 7, 2g):
Wenn wir erstmalig unserer Gebrauchsanweisungen auf unserer Website zur Verfügung stellen, müssen wir dann auch frühere Versionen hochladen? Bezieht sich der Passus „alle früheren Versionen der in elektronischer Form herausgegebenen Gebrauchsanweisung“ auf die bisherigen Versionen auf der Website?
Herzlichen Dank und viele Grüße
Heike Hauptmann
Sehr geehrte Frau Hauptmann,
als Experte für Gebrauchsanweisungen beantworte ich gerne Ihre Frage.
Sie müssen nur die Versionen der Gebrauchsanweisung auf Ihre Website stellen, welche bereits elektronisch Inverkehr gebracht wurden. Haben Sie beispielsweise bereits elektronische Versionen der IFU auf einer CD oder über ein Display Ihres Produkts oder auf einer anderen Website Inverkehr gebracht, müssen Sie diese älteren elektronischen Versionen auch auf Ihrer Website zur Verfügung stellen. Versionen, die ausschließlich in Papierform Inverkehr gebracht wurden, müssen nicht auf die Website gestellt werden.
Ich hoffe damit beantworte ich Ihre Frage. Ansonsten haken Sie gerne nach.
Beste Grüße
Nils Becker
Sehr geehrter Herr Becker,
haben Sie recht herzlichen Dank für Ihre Antwort, die für uns so ausreichend ist.
Wir hätten noch eine weitere Frage in diesem Zusammenhang.
Die Anwender unserer Medizinprodukte sind ausschließlich medizinisches Fachpersonal.
Wie muss die Zugehörigkeit zu dieser Gruppe auf der Internetseite bestätigt werden bevor die elektronischen GAs eingesehen werden können? Kann das durch Bestätigung eines Buttons erfolgen und dann sind die GAs verfügbar oder dürfen die Benutzer nur über einen Login Zugang bekommen?
Herzlichen Dank und viele Grüße
Heike Hauptmann
Sehr geehrte Frau Hauptmann,
mir ist keine Forderungen bekannt, die besagt, dass die Zugehörigkeit der Nutzergruppe vor Einsicht der eIFU auf der Website bestätigt werden muss. Falls ich was übersehen haben sollte, wäre ich Ihnen dankbar für diesen Hinweis.
Herzliche Grüße
Nils Becker
Zitat der o.g. Kardinalfehler bei IFUs:
* Sie stimmt nicht (mehr) mit dem Produkt überein (das ist besonders bei SW-Updates der Fall).
* Es ist unklar, auf welches Produkt bzw. auf welche Variante sich die Gebrauchsanweisung bezieht und welche Version der Gebrauchsanweisung vorliegt.
Ich finde bisher keine klare normative oder gesetzliche Vorgabe für diese banale und völlig vernünftige Pflicht:
Wie stellt der Hersteller eines PEMS sicher, dass der Anwender sofort erkennen kann, ob vorliegende IFU/Gebrauchsanweisung zum vorgestellten Gerät und seiner Software-Version wirklich passt?
Ich habe das m.E. dadurch einwandfrei gelöst, indem die Geräte-SW z.B. eine „IFU: 13.x“ im Einschaltbild verlangt und dazu die IFUs V 13.0 bis 13.99 passen. – Die IFU V 14.0 wäre demnach einfach erkennbar inkompatibel – und enthielte zusätzlich noch den Hinweis, ab welcher SW-Version genau sie gilt.
Soweit so gut – aber ständig droht das Scharmützel mit den Minimalisten, die das immer wieder zu unterlaufen suchen…
Deswegen muss ich eine verpflichtende (ggf. auch internationale) Vorgabe für die gegenseitig eindeutige Kompatibilität Geräte-SW zu den IFU finden.
Besten Dank
Frank Frankus
Sehr geehrter Herr Becker,
meine eigentliche Frage ging vielleicht unter:
Wo ist normativ festgelegt, dass man direkt erkennen kann, ob vorliegende Paarung aus IFU und Geräte(-SW-Version) wirklich zusammengehören?
Ich hätte das in der Grundnorm 60601 erwartet, konnte aber weder hier noch in den anderen Zitaten das so explizit finden.
Besten Dank,
Frank Frankus
Sehr geehrter Herr Frankus,
die Anforderung, dass die IFU mit dem Produkt ( bzw. der Geräte-SW) übereinstimmen muss, findet sich in der MDR.
In Artikel 7 heißt es, dass es untersagt ist, in der Gebrauchsanweisung „dem Produkt Funktionen und Eigenschaften zuschreiben, die es nicht besitzt“.
In Anhang I 23.4 a wird die Angabe des „Namen oder Handelsnamen des Produkts“ gefordert, wozu ggf. auch die Versionsnummer zählt. Ferner wird unter Punkt f gefordert, dass die Gebrauchsanweisung Angaben enthält, „anhand deren ein Angehöriger der Gesundheitsberufe überprüfen kann, ob das Produkt geeignet ist, und die entsprechende Software und die entsprechenden Zubehörteile auswählen kann;“.
Ganz konkret wird in der IEC/IEEE 82079-1:2019 unter Kapitel 7.2 gefordert, dass in der Gebrauchsanweisung angegeben wird, für welche Versionsnummern des Produkts die Gebrauchsanweisung gilt. Die 82079-1 ist allerdings keine unter MDD/MDR harmonisierte Norm.
Spätestens nach einer Risikoanalyse nach 14971 sollte dem Hersteller klar werden, dass die fehlende Angabe des Produktegeltungsbereichs in der Gebrauchsanweisung zu Risiken führen könnte, insbesondere bei mehreren Produktversionen.
Ich hoffe, meine Antwort hilft Ihnen weiter. Falls nicht, fragen Sie gerne nach.
Herzliche Grüße
Nils Becker
Liebes Johner-Team,
die EN 1041:2008 wird als harmonisierte Norm unter MDD geführt. Laut Angabe in der DIN EN 1041:2013-12, Anhang ZA soll die Norm aber „nicht als Mittel zur Sicherstellung der Einhaltung der Anforderungen der Medizinproduktrichtlinien verwendet werden“.
Wie ist damit im Hinblick auf die MDR umzugehen? Welche Bedeutung kommt der EN 1041 unter der MDR zu?
Vielen Dank vorab und beste Grüße
Sonja Oechsler
Liebe Frau Oechsler,
besten Dank für Ihre Frage! Die ISO 1041:2013 schreibt im ZA Anhang, dass diese Norm nicht als Checkliste für die Einhaltung der Anforderungen der MDD genutzt werden soll, da „es mehrere Unstimmigkeiten bezüglich des Inhalts von Anhang A [der ISO 1041] und den Anforderungen in dem
ersten Anhang jeder relevanten Medizinprodukterichtlinie [MDD, 93/42/EWG]“ gibt. Zwischen der MDR und ISO 1041 kommen noch weitere Unstimmigkeiten hinzu.
Die Autoren der ISO 1041 sind sich dieser Unstimmigkeiten bewusst und möchten die Norm deshalb in Bezug auf die MDR anpassen. Die ISO 1041 soll durch die ISO 20417 ersetzt werden, welche unter der MDR harmonisiert werden soll. Bisher liegt die ISO 20417 allerdings nur als Entwurf vor. Somit gilt die ISO:1041 weiterhin als Stand der Technik und sollte unter MDD und MDR wo anwendbar umgesetzt werden. Zusätzlich müssen alle Anforderungen der MDR aus Anhang I Kapitel 23 erfüllt werden.
Ich hoffe, meine Antwort hilft Ihnen weiter.
Herzliche Grüße, Nils Becker
Liebes Johner-Team,
wir sind ein Großhandel und vertreiben zum Großteil MP. Ist es für uns Pflicht, die Sprachen der mitgelieferten Gebrauchsanweisung und der Produktetiketten zu hinterlegen?
Vielen Dank für Ihre Frage!
Die MDR stellt unter Artikel 14 allgemeine Pflichten an den Händler. Zu diesen Pflichten zählt die Überprüfung, ob alle geforderten Angaben in der Gebrauchsanweisung und in der Kennzeichnung gemacht wurden und ob diese in der jeweils geforderten Landessprache des Landes vorliegen, in welches Sie das Produkt vertreiben.
Ich hoffe , dass ich Ihre Frage auch richtig verstanden habe. Wenn nicht, haken Sie gerne nach.
Herzliche Grüße
Nils Becker
Hallo liebes Johner-Team,
in welche Sprachen muss ich denn eine Gebrauchsanweisung übersetzen, wenn ich in ein mehrsprachiges Land verkaufen möchte? Zum Beispiel Belgien oder Schweiz.
Wo finde ich die Info, dass man beispielsweise für die Schweiz alle drei Sprachen (deutsch, französisch, italienisch) benötigt. Gibt es eine Verordnung bzgl. dieser Sprachregelung?
Viele Grüße
Lieber Herr Bahl,
die MDR schreibt in Artikel 10 Absatz 11, dass die Gebrauchsanweisung „in einer oder mehreren von dem Mitgliedstaat, in dem das Produkt dem Anwender oder Patienten zur Verfügung gestellt wird, festgelegten Amtssprache(n) beiliegen“ muss. Jeder EU-Mitgliedsstaat legt die Sprachanforderungen selber in nationalen Gesetzen oder Verordnungen fest. In Deutschland wird das beispielsweise im Medizinprodukte-EU-Anpassungsgesetz–MPEUAnpG festgelegt, in Belgien im „Arrêté royal relatif aux dispositifs médicaux, 1999-03-18“.
Wir haben in mühevoller Recherche die Sprachanforderung aller EU-Mitgliedsstaaten in einer Liste zusammengetragen. Bei Interesse an dieser Liste nehmen Sie gerne Kontakt mit uns auf.
Herzliche Grüße
Nils Becker
Sehr geehrter Herr Becker,
wenn ich als Hersteller bei einer Risikoanalyse feststellen sollte, daß eine äußerlich den Thorax abstützende/umformende Thoraxorthese keinesfalls in Gegenwart eines innerlich den Thorax abstützenden/umformenden Implantats angewandt werden soll/darf, wo vermerke ich das zweckmäßigerweise in den Gebrauchsanweisungen?
A) in der Nähe der Zweckbestimmung in den Einschränkungen zum Bestimmungsgemäßen Gebrauch?
B) Bei den Kontraindikationen?
C) Bei den Kritischen Warnungen
Falls möglich die Antwort bitte basierend auf einer Begriffsabgrenzung von
a) (Einschränkungen zum) Bestimmungsgemäßen Gebrauch?
b) Kontraindikationen
c) Kritische Warnungen
Vielen Dank im Voraus.
Eckart Klobe
Sehr geehrter Herr Klobe,
vielen Dank für Ihre Frage.
Die MDR oder die unter der MDD harmonisierten Normen machen keine Vorgaben zur Platzierung von sicherheitsbezogenen Informationen in der Gebrauchsanweisung.
Hier lohnt sich ein Blick in die IEC/IEEE 82079-1, welche als Stand der Technik für das Erstellen von Gebrauchsanweisungen angesehen werden kann. Diese schreibt, dass Sicherheitshinweise gruppiert am Anfang der Gebrauchsanweisung platziert werden sollen. Warnhinweise sollen in die Schritt-für-Schritt-Anleitung an der Stelle integriert werden, wo das Auftreten einer entsprechenden Gefährdung erwartet wird.
Die Sicherheitsinformation, die Sie beschreiben, würde demnach gut in alle 3 Möglichkeiten A, B und C passen. Die wichtigste Überlegung bei Ihrer Entscheidung sollte sein, dass die vorgesehenen Anwender die Information erstens entdecken und zweitens verstehen (das sollte im Rahmen eines kleinen Usability-Tests der GA mit vorgesehenen Anwendern überprüft werden). Es spricht auch nichts dagegen, essenzielle Sicherheitsinformationen mehrfach in der Gebrauchsanweisung aufzuführen.
Auch wenn ich nicht sagen kann, ob Sie sich für Option A, B oder C entscheiden sollten, hoffe ich meine Antwort hilft Ihnen weiter.
Herzliche Grüße
Nils Becker
Sehr geehrter Herr Becker,
in Kapitel 2 e) dieses Blogs haben Sie dankenswerterweise einen Link zu einer Auflistung gelegt, in welcher zusammengetragen wurde, in welchem europäischen Land die Gebrauchsanweisung in welcher Sprache nach 93/42/EEC beigegeben werden muss bzw. darf.
Meine Fragen:
1) Sind nach MDR diesbezüglich Änderungen zu erwarten? Falls JA, was sind die wesentlichsten?
2) Könnten Sie noch eine Auflistung der nach Norm XY (??) zulässigen (oder lediglich empfohlenen?) Abkürzungen der entsprechenden Sprachen in der Kennzeichnung der verschiedenen Sprachen-Versionen der Gebrauchsanweisungen nachliefern oder gar in Ihre oben genannte Auflistung mit einarbeiten?
Vielen Dank im Voraus.
Eckart Klobe
Sehr geehrter Herr Klobe,
vielen Dank für Ihre beiden Fragen, auf die ich sehr gerne eingehe.
1) In welcher Sprache die IFU vorliegen muss, ist in der nationalen Gesetzgebung des jeweiligen Landes geregelt. Grundsätzlich muss die IFU immer in der jeweiligen Landessprache des Landes, in dem das Produkt vertrieben wird, vorliegen. In Sonderfällen, z.B. wenn die vorgesehenen Nutzer professionelle Nutzer sind, sind landesspezifische Ausnahmen möglich. Dies galt auch unter der MDD schon. Im Zuge der Umstellung auf die MDR haben vereinzelt Länder ihre Sprachanforderungen geändert oder präzisiert. Gerne können wir Ihnen eine Auflistung, in welchem EU-Land welche Sprachen aktuell vorgeschrieben sind, anbieten. Da die Recherche dazu recht umfangreich war und wir die Informationen kontinuierlich aktualisieren, können wir diese Liste leider nicht kostenlos anbieten.
2) In der ISO/FDIS 20417:2020, welche unter der MDR harmonisiert werden und die EN 1041 ersetzen soll, werden folgende Normen zur Angabe der Sprachcodes aufgelistet:
ISO 639-1, Codes for the representation of names of languages — Part 1: Alpha-2 code
ISO 639-2, Codes for the representation of names of languages — Part 2: Alpha-3 code
ISO 639-3, Codes for the representation of names of languages — Part 3: Alpha-3 code for comprehensive coverage of languages
Die Angabe der Sprachcodes kann, wie in den Normen spezifiziert, demnach entweder mit 2 oder 3 Buchstaben gemacht werden.
Herzliche Grüße
Nils Becker
Liebes Johner Team,
wir sind Hersteller von Implantaten (Klasse III). In Anhang I 23.4 (aa) MDR heißt es: „Die Gebrauchsanweisung enthält alle folgenden Angaben: Patienten mit einem implantierten Produkt gemäß Artikel 18 zur Verfügung zu stellende Informationen“. Gem. Artikel 18 gehören zu diesen Informationen u.a. die UDI, Seriennummer und LOT.
Bedeutet dies tatsächlich, dass wir in der IFU die UDI (inkl. PI) bzw. die LOT aller Produkte, die in den Scope der IFU fallen, aufnehmen müssen?
Vielen Dank im Voraus und viele Grüße
Peter Koszolka
Lieber Herr Koszolka,
Informationen wie die UDI-PI und LOT müssen gemäß Anhang I 23.4 (aa) generell zur Verfügung gestellt werden. Gemäß Artikel 18 sollen derartige Informationen über den Implantationsausweis zur Verfügung gestellt werden. Dies können Sie beispielsweise in Form von Abreißetiketten auf dem Implantat, welche in einen Implantationsausweis geklebt werden können, oder durch einen vorausgefüllten Ausweis für den Patienten tun. Weiterführende Informationen hierzu finden sie hier:
https://ec.europa.eu/docsroom/documents/40321/attachments/1/translations/en/renditions/native
Liebe Grüße
Philipp Schleer
Hallo liebes Johner-Team,
wenn ich auf Basis einer Gebrauchsanweisung für ein (Medizin)Produkt in der EU (Gebrauchsanweisung enthält das CE-Zeichen) nun eine separate Gebrauchsanweisung für den US-Markt erstelle, muss dann zwingend das CE-Zeichen entfernt werden, da es nur innerhalb der EU gilt oder könnte es dennoch in der US-Gebrauchsanweisung auftauchen?
Freundliche Grüße
Marius
Lieber Marius,
solange die US-Gebrauchsanweisung identisch zur Gebrauchsanweisung in der EU ist und es sich dabei lediglich um eine Übersetzung, am besten von einem zertifizierten Übersetzer, handelt, darf diese das CE Kennzeichen tragen, obwohl es auf dem US-Markt keine Bedeutung hat. Sollte die Gebrauchsanweisung jedoch speziell für den US-Markt erstellt worden sein und nicht identisch zur EU-Variante sein, muss das CE Zeichen entfernt werden, da sie ein Teil des Produktes ist und das Produkt nur das CE-Zeichen tragen darf solange alle Teile identisch sind.
Liebe Grüße
Philipp Schleer
Sehr geehrter Herr Becker,
wenn Produkte aus dem Portfolio genommen werden und beispielsweise zum 31.12.2021 auslaufen sollen und für die verbleibenden Produkte die Gebrauchsanweisung nachgedruckt werden soll, können die Auslaufprodukte aus der IFU herausgenommen werden (die Auslaufprodukte sind aber noch in verschiedenen Lägern verfügbar, bis der Bestand auch erschöpft ist)?
Oder müssen die Produkte solange in der Gebrauchsanweisung aufgelistet sein, bis auch das letzte Produkt verkauft ist?
Ich hoffe, dass ich mein Anliegen einigermaßen verständlich beschreiben konnte.
Vielen dank für Ihre Hilfe!
Mit freundlichen Grüßen
Christine Graß
Sehr geehrte Frau Graß,
ich bin mir nicht sicher, ob ich Ihre frage korrekt verstanden habe, aber gerne versuche ich eine Antwort.
Laut ISO 20417 Kapitel 6.6.1 (3) muss dass Produkt, für welches die Gebrauchsanweisung gilt, in der Gebrauchsanweisung klar aufgeführt werden. Entweder durch den kommerziellen Produktnamen, den Namen der Medizinproduktefamilie, der Modell- oder Katalognummer.
Wenn Ihre neugedruckten Gebrauchsanweisungen also auch in Zukunft noch für die gelagerten aber aus dem Portfolio genommen Produkte dienen soll, müssen diese Produkte auch klar in der neuen Gebrauchsanweisung aufgeführt werden.
Ich hoffe, ich konnte Ihnen mit meiner Antwort weiterhelfen.
Herzliche Grüße
Nils Becker
Sehr geehrter Herr Becker,
ja, sie haben meine Frage richtig verstanden.
Danke für Ihre hilfreiche Antwort. Sie haben damit mein „Bauchgefühl“ bestätigt.
Mit freundlichen Grüßen
Christine Graß
Sehr geehrte Damen und Herren,
es gibt immer wieder Diskussionen ob ein Service-Manual (also weniger für die Anwender sondern für die Medizintechniker zur Instandhaltung) vom Hersteller mitzuliefern ist. Unserer Ansicht wäre das für die Erfüllung der MPBetreibV für unsere internen Mitarbeiter wichtig und erforderlich.
Leider ist kaum ein Hersteller dazu zu bewegen. Gibt es hierzu keine Vorgaben ?
Sehr geehrter Herr Tuma,
besten Dank für Ihre Frage.
Es gibt keine regulatorischen Anforderungen, dass ein eigenständiges, separates „Service-Manual“ dem Produkt beigelegt werden muss.
Sehr wohl fordert die MDR aber, dass in der Gebrauchsanweisung dem Anwender (oder dritten Personen) alle relevante Informationen über Sicherheit und Leistung mitgeteilt werden. Dazu zählt ggf. auch die Instandhaltung. Explizit fordert die MDR in Anhang I Absatz 23.4 i und k Angaben zur Endmontage, Installation und Instandhaltung. Beispielweise heißt es, dass alle „Angaben zur Art und Häufigkeit präventiver und regelmäßiger Instandhaltungsmaßnahmen sowie zur eventuellen vorbereitenden Reinigung oder Desinfektion“ in der Gebrauchsanweisung gemacht werden müssen.
Diese Angaben können in einem extra „Service Manual“ gemacht werden, müssen aber nicht.
Ich hoffe, meine Antwort hilft Ihnen weiter. Ansonsten haken Sie gerne nach.
Herzliche Grüße
Nils Becker
Hallo Herr Professor Johner,
vielen Dank für die Informationen und die Auflistung der Sprachen.
Auf der EU-Website habe ich leider keine Liste mit den Sprachen gefunden.
Im EU-Register habe ich nach den Amtssprachen geschaut, aber diese lässt sich ja nicht auf die IFUs und andere TD anwenden, denn Gälisch brauchen wir nicht:
https://eur-lex.europa.eu/legal-content/DE/TXT/?uri=CELEX%3A32017R0745
Ihre obige Liste ist aus dem Jahr 2008:
https://www.johner-institut.de/blog/wp-content/uploads/2018/01/MDEG_-_2008-12_-_II-6.3._Mandatory_Languages_Requirements_for_Medical_Devices_update_Sept.08.pdf
Hat sich bei der MDR etwas geändert?
Übrigens, auf Ihrer Website ist noch eine weitere Liste (aus dem Jahr 2004, da fehlt z. B. Bulgarien, war damals ja noch nicht Mitglied):
https://www.johner-institut.de/blog/wp-content/uploads/2015/10/020205_Mandatory_Language.pdf
Sind die Sprachenkürzel dann nach der ISO 639-1?
Vielen Dank.
Mit freundlichen Grüßen
Ayfer Bektas
Liebe Frau Bektas,
besten Dank für Ihre Fragen und wertvollen Hinweise.
Leider gibt es nach meinem Kenntnisstand keine aktuelle, offizielle und öffentliche Liste, die die Sprachanforderung der EU-Mitgliedsstaaten an die IFU und Kennzeichnung auflistet. Wir führen intern eine entsprechende Liste und wenn Sie daran Interesse haben, melden Sie sich gerne bei uns über das Formular.
Prinzipiell hat die MDR nichts an den Sprachanforderungen geändert. Auch die MDR schreibt in Artikel 10 (11):
„Die Hersteller sorgen dafür, dass dem Produkt die Informationen gemäß Anhang I Abschnitt 23 in einer oder mehreren von dem Mitgliedstaat, in dem das Produkt dem Anwender oder Patienten zur Verfügung gestellt wird, festgelegten Amtssprache(n) der Union beiliegen.“
Es bleibt also Sache der einzelnen Mitgliedsstaaten, die Sprachanforderungen festzulegen.
Wenn der Hersteller Begleitinformationen, wie die IFU, mehrsprachig bereitstellt, sollten die jeweiligen Sprachen als Text identifizierbar sein oder es sollten die Kürzel nach ISO 639-1, ISO 639-2 oder ISO 639-3 verwendet werden. Das ist aber eine „Sollte“- und keine „Muss“-Anforderung aus der ISO 20417 Kapitel 5.3.1.
Ich hoffe meine Antwort hilft Ihnen weiter, ansonsten haken Sie gerne nach.
Herzliche Grüße
Nils Becker
PS: Ich darf Ihnen herzliche Grüße von Herrn Johner ausrichten.
Liebes Johner-Team,
wir legen einem Klasse IIa Medizinprodukt eine grafische IFU bei, in welcher wir auch auf unser Website verweisen, wo die Text-IFU in unterschiedlichen Sprachen heruntergeladen werden kann.
Bei der grafischen IFU haben wir bei einem Audit der benannten Stellen einen Hinweis bekommen, dass für jedes Land, in dem das Produkt verkauft wird, in der grafischen IFU Kontaktdaten des jeweiligen Landes angegeben werden müssen. Ist das richtig? Wenn ja, wo ist diese Anforderung definiert?
Beste Grüße,
Martina
Liebe Frau Marker,
besten Dank für Ihre Frage.
Ich bin zugegeben etwas überrascht, denn laut MDR muss die IFU und alle darin geforderten Angaben (siehe MDR Anhang I, Absatz 23.4) in Papierform beigelegt werden (siehe MDR, Anhang I, Absatz 23.1 f). eIFUs sind unter bestimmten Bedingungen möglich (siehe https://www.johner-institut.de/blog/regulatory-affairs/elektronische-gebrauchsanweisung-medizinprodukte-207-2012/).
Ob nun als Papier-IFU oder eIFU, der Hersteller muss „die Anschrift seiner eingetragenen Niederlassung“ angeben (siehe MDR Anhang I, Absatz 23.4 a). Die ISO 20417 fordert zudem in 6.6.1 c 2, dass die Kontaktdaten (z.B. Telefonnummer, Webseite, Email-Adresse, etc. ) unter denen man technische Hilfe bekommen kann, angegeben werden müssen.
Herzliche Grüße
Nils Becker
Hallo,
bezugnehmend auf die Frage von Martina vom 30.01.2022 habe ich noch den Hinweis, dass es für IVDs die MEDDEV 2.14/3 gibt. Diese gibt sehr wohl vor, dass u.a. entsprechende lokale Vertriebsniederlassung, etc. anzugeben ist.
Wir lösen dies so, dass wir ein zusätzliches Dokument mit den entsprechenden Länderspezifischen Daten dem Produkt beilegen.
Ob es ein vergleichbares Dokument für Medizinprodukte gibt, weiß ich leider nicht. Eventuell hat das Johner-Team hierzu noch weitere Informationen.
Beste Grüße,
Susanne
Liebe Susanne,
vielen Dank für Ihren wertvollen Hinweis. In der MEDDEV 2.14/3 wird in Kapitel 5.2 folgende Information in der eIFU gefordert:
„The name and address of the manufacturer or authorized representative“
Ein vergleichbares MEDDEV Dokument für eIFUs für Medizinprodukte gibt es nicht. In dem Zusammenhang ist vor allem die EU VO 2021/2226 wichtig (https://eur-lex.europa.eu/legal-content/DE/TXT/HTML/?uri=CELEX:32021R2226&from=DE).
Herzliche Grüße
Nils Becker
Lieber Christian,
du schreibst in deinem Blog zum Thema Gebrauchsanweisungen unter:
„2. Regulatorische Anforderungen an Gebrauchsanweisungen, g) Anforderungen an die Übersetzung der Gebrauchsanweisungen“,
dass die MDR keine expliziten Anforderungen an die Übersetzung der Gebrauchsanweisung stellt. Nachfolgend zählst du einige Kompetenzen auf, die der Übersetzer haben „sollte“, aber offensichtlich nicht „muss“.
Heißt das nun, dass es regulatorisch korrekt wäre, wenn die Übersetzungen inhouse von einem Muttersprachler übernommen werden, da die MDR ein zertifiziertes Übersetzer nicht explizit vorschreibt, oder bin ich verpflichtet die von dir zitierten Regularien
„ISO 17100:2015“ und
„IEC/IEEE 82079‑1“ einzuhalten, damit ich den „State of the art“ einhalte?
Ich danke dir sehr für eine Antwort.
Herzliche Grüße
Petra
Liebe Petra,
gerne antworte ich an Christians Stelle.
Die Regularien ISO 17100:2051 und IEC/IEEE 82079‑1 haben keinen Branchenfokus. Sie sind nicht verpflichtend für Medizinproduktehersteller einzuhalten, zumindest ist mir bisher keine derartige Forderung bekannt. Bei geeigneter Kompetenz kann die Übersetzung also auch „inhouse“ vorgenommen werden. Unbedingt ist dabei zu beachten, dass die Qualität der Übersetzung und somit die Verständlichkeit der Gebrauchsanweisung gewahrt wird. Das Gegenteil kann zu ernsthaften Risiken führen.
Ich hoffe meine Antwort hilft Dir weiter, ansonsten frag gerne nach.
Herzliche Grüße
Nils
Lieber Nils,
herzliche Dank für deine schnelle Antwort. Nun hat sich noch eine weitere Frage ergeben. Ist das auch für die Mehrsprachigkeit von Software anzuwenden? Kann das auch von einer geeigneten Person „inhouse“ erledigt werden?
Herzliche Grüße
Petra
Liebe Petra,
auch bezüglich der Übersetzung der GUI stellt die MDR keine expliziten Anforderungen. Auch hier muss allerdings gewährleistet sein, dass die Übersetzung die Usability nicht gefährdet.
Herzliche Grüße
Nils
Wir vertreiben IVD Diagnostica, auch ein Gerät für die Messung von Blut in Stuhlproben. Wie sind hier die Regularien. Ist eine deutsche Menüführung zwingend vorgeschrieben.
Oder gibt es hier auch Ausnahmen wie bei der Gebrauchsanweisung.
Über Hinweise wo ich hierzu Informationen finden könnte, wäre ich Ihnen dankbar.
Herzliche Grüße
Johannes Reinholz
Sehr geehrter Herr Reinholz,
besten Dank für Ihre Frage!
Die Sprachanforderungen, auch an das Graphical User Interface, regelt jedes EU-Mitgliedsland selbst. In Deutschland z. B. regelt das Medizinprodukterecht-Durchführungsgesetz – MPDG die Sache. In § 8 heißt es:
„(2) Produkte dürfen im Geltungsbereich dieses Gesetzes nur dann an Anwender und Patienten abgegeben werden, wenn die für Anwender und Patienten bestimmten Informationen in deutscher Sprache zur Verfügung gestellt werden. In begründeten Fällen dürfen die Informationen auch in englischer Sprache oder einer anderen für den Anwender des Medizinproduktes leicht verständlichen Sprache zur Verfügung gestellt werden, wenn diese Informationen ausschließlich für professionelle Anwender bestimmt sind und die sicherheitsbezogenen Informationen auch in deutscher Sprache oder in der Sprache des Anwenders zur Verfügung gestellt werden.“
Unter „Informationen“ sind auch Informationen auf der Benutzeroberfläche der Software zu verstehen. Diese müssten also in Deutsch sein. Ausnahme: für Angehörige der Gesundheitsberufe (professionelle Anwender) ist Englisch auch erlaubt, vorausgesetzt sicherheitsbezogene Informationen sind auf Deutsch.
Viele weitere EU-Mitgliedsländer haben ähnliche Sprachregelungen.
Zudem muss der Hersteller im Rahmen des Risikomanagement plausibel darlegen können, dass kein inakzeptables Risiko entsteht, wenn die Informationen auf Englisch sind.
Ich hoffe meine Antwort hilft Ihnen weiter.
Herzliche Grüße
Nils Becker
Hallo Herr Prof Johner, hallo Herr Dr. Becker,
welche Unterlagen sind, Ihrer Ansicht nach, verpflichtend in die jeweiligen Landessprachen zu übersetzen?
Folgende:
Gebrauchsanweisung, Labeling (Etiketten), Konformitätserklärung, SPUR, Benutzeroberfläche bei Software, Field Safety Notice?
Vielen Dank für Ihre Hilfe.
Viele Grüße
C.Kalle
Lieber Herr Kalle,
herzlichen Dank für Ihre Frage.
Gebrauchsanweisung sowie Kennzeichnung müssen nach Artikel 10 (11) der MDR „in einer oder mehreren von dem Mitgliedstaat, in dem das Produkt dem Anwender oder Patienten zur Verfügung gestellt wird, festgelegten Amtssprache(n) der Union beiliegen.“
Die gilt in der Regel auch für Informationen auf der Benutzeroberfläche (der Software), falls nicht anders von dem jeweiligen Mitgliedsstaat festgelegt.
„Die Sicherheitsanweisung im Feld [Field Safety Notice] ist in einer Amtssprache oder in Amtssprachen der Union abzufassen, entsprechend der Vorgabe durch den Mitgliedstaat, in dem die Sicherheitskorrekturmaßnahmen im Feld ergriffen werden.“ Siehe MDR Artikel 89 (8).
Die EU-Konformitätserklärung „wird in eine oder mehrere Amtssprachen der Union übersetzt, die von dem/ den Mitgliedstaat(en) vorgeschrieben wird/werden, in dem/denen das Produkt bereitgestellt wird.“ Siehe MDR Artikel 19.
Der PSUR gemäß MDR Artikel 86 unterliegt keinen Bestimmungen bezüglich der Sprache. Der Kurzbericht über Sicherheit und klinische Leistung (siehe Artikel 32 der MDR) ist für die Öffentlichkeit zugänglich und sollte demnach ebenfalls in die von dem jeweiligen Mitgliedsatt gefordert Sprachen übersetzt werden.
Ich hoffe meine Antwort hilft Ihnen weiter.
Herzliche Grüße
Nils Becker
Lieber Herr Becker,
allerbesten Dank für die schnelle und kompetente Rückmeldung.
Ist verpflichtend die Homepage in den jeweiligen Amtssprachen der Mitgliedstaaten zur Verfügung zu haben? (Hinweis: Auf der Homepage sind keine e-IFUs oder e-Kataloge abrufbar)
Ein Diskurs, den wir derzeit auch mit einem Berater führen:
Marketingmaterial (z.B. Broschüren, Präsentationen), das nicht im Lieferumfang enthalten ist, soll tatsächlich in die Amtssprachen der Mitgliedsstaaten, in die wir liefern, übersetzt werden?
Mit mehr Fragen werde ich Sie nicht strapazieren und
bedanke mich schon im Voraus für Ihre Geduld & Hilfe 🙂
Viele Grüße
C.Kalle
Lieber Herr Kalle,
vielen Dank für Ihre Nachfrage, die ich ebenfalls gerne beantworte.
Einige Länder, wie z.B. Spanien, Italien und die USA, fordern tatsächlich die Übersetzung der Website und des Werbematerials. Sie sollten also unbedingt die nationalen Vorgaben der Länder, in die Sie Ihr Produkt Inverkehr bringen möchten, prüfen.
Herzliche Grüße
Nils Becker
Sehr geehrter Herr Johner,
Gibt es Vorschriften, wie die Literaturangaben in der Gebrauchsanweisung zu gestalten sind.
Konkret gefragt:
Wir möchten gerne eine sehr neues Studienergebnis einfügen. Es wurde bisher aber erst ein Poster auf einem Kongress veröffentlicht. Und dazu gibt es keine öffentlichen Zugang (z.B. Abstraktband, etc.).
Vielen Dank für Ihre Hilfe!
Beste Grüße,
P. Zürbig
Sehr geehrte Frau Zürbig,
vielen Dank für Ihre Frage. Gerne antworte ich an Herrn Johners Stelle.
Die ISO 18113-2 verlangt, dass „Einschlägige Literaturangaben“ anzugeben sind. Es gibt jedoch keine konkreten Anforderungen, wie die Literaturangaben zu gestalten sind.
Das Ziel der Gebrauchsanweisung und der darin enthaltenen Literaturangaben ist es, dem Anwender wichtige Informationen zur Verfügung zu stellen. Wichtige Literatur sollte meiner Einschätzung nach also öffentlich zugänglich sein, was bei einem Poster eher nicht der Fall ist.
Ich hoffe meine Antwort hilft Ihnen weiter, ansonsten haken Sie gerne nach.
Herzliche Grüße
Nils Becker
Sehr geehrter Herr Johner,
kann der Hersteller [M] (außerhalb EU ansässig, Bevollmächtigter vorhanden) uns als Importeur und Distributor [I/D]
1. die Übersetzung aufzwingen? „I/D will provide M with instructions for use in German in an electronic format.“
2. die Kosten für weitere Übersetzungen aufzwingen? „Translations of instructions for use (and any related essential literature) into languages other than English will be at I/D’s expense.“
Vielen herzlichen Dank,
K. Münzner
Sehr geehrte Frau Münzner,
vielen Dank für Ihre Frage, die ich gerne an Herrn Johners Stelle beantworte.
Labeling (IFU) inkl. Übersetzung liegt in der Verantwortung des Herstellers, soweit keine individuellen anderen Vereinbarungen getroffen wurden. Der Hersteller muss das Produkt mit allen Informationen in der vom Zielmarkt festgelegten Sprache zu liefern. Die Übersetzung kann ausgelagert werden, ist dann aber ein ausgelagerter Prozess und muss entsprechend überwacht werden (z.B. Freigabe jeder Version der Übersetzung durch den Hersteller). Es gibt keine regulatorische Grundlage, auf der sich diese Verantwortung oder damit verbundene Kosten auf den Importeuer aufzwingen lassen, es sei denn, es gibt eine entsprechende Vereinbarung zwischen Hersteller und Importeur.
Ich hoffe, meine Antwort hilft Ihnen weiter. Ansonsten haken Sie gerne nach.
Herzliche Grüße
Nils Becker
Liebes Johner Team,
wir haben ein Medizinprodukt der Klasse I. Ist der Vigilanz-Hinweis nach GSLA 23.4.z zwingend erforderlicher Bestandteil einer jeden Gebrauchsanweisung (sofern vorhanden), für alle Klassen, oder kann hier ein Ausschluss definiert werden?
Gruß Walter
Lieber Herr Walter,
die Meldepflicht von schwerwiegenden Vorkommnissen ist eine grundlegende Forderung der MDR hinsichtlich des Vigilanz-Systems. Die Forderung über einen entsprechenden Hinweis in der Gebrauchsanweisung ist unabhängig von der Risikoklasse und für alle Produkte gleichermaßen verpflichtend.
Liebe Grüße
Philipp Schleer
Lieber Herr Schleer,
vielen Dank für die schnelle Antwort. Super! Gestatten Sie trotzdem noch die Nachfrage. Die MDR macht hier keine Ausnahmen in den Klassen? Ich will verdeutlichen und frage hinsichtlich der Verhältnismäßigkeit: Ein Pflaster als Medizinprodukt der Klasse I wird kaum ein Vorkommnis auslösen.
Danke für eine Klarstellung im Voraus.
Herzliche Grüße Dr. Walter
Lieber Dr. Walter,
das ist korrekt, die MDR macht hier keine Ausnahmen in den Klassen. Auch bei einem Pflaster ist rein theoretisch eine allergische Reaktion denkbar, welche gegebenenfalls meldepflichtig sein könnte. Die einzige Möglichkeit den Hinweis in der Gebrauchsanweisung wegzulassen wäre, wenn absolut ausgeschlossen ist, dass jemals ein schwerwiegendes Vorkommnis vorkommen kann. Den Nachweis hierfür müsste man dann über das Risikomanagement erbringen, allerdings ist dies extrem schwierig, weswegen ich Ihnen empfehlen würde den Hinweis aufzunehmen.
Liebe Grüße
Philipp Schleer
Liebes Johner Team,
die Norm DIN EN ISO 15223-1:2022-02 beschreibt in der Tabelle der Symbole ein Symbol, dass die Gebrauchsanweisung zu beachten ist (Bezugsnummer 5.4.3). Gleichzeitig wird jedoch auch bei den Anmerkungen der Hinweis auf ein anderes Symbol mit derselben Bedeutung gegeben. Dabei handelt es sich um das Sicherheitszeichen ISO 7010-M002.
Jetzt stellt sich mir die Frage: Bei zusätzlicher Betrachtung der ISO 20417 erscheint es mir, als wäre es mir selbst überlassen, welches Symbol ich gerne verwenden möchte. Stimmt das, oder ist meine Annahme falsch?
Vielen Dank für Ihre Unterstützung bereits im Voraus!
Herzliche Grüße
Dennis Schall
Lieber Herr Schall,
herzlichen Dank für Ihre Frage.
Die ISO 20417 unterscheidet tatsächlich die Verwendung der Symbole 5.4.3 der ISO 15223-1 und ISO 7010-M002. ISO 7010-M002 ist zu verwenden, wenn das Lesen der IFU als Risikokontrollmaßnahme (RKM) für ein bestimmtes Risiko definiert ist. Stellt die IFU keine RKM dar, kann (muss aber nicht) das Symbol 5.4.3 verwendet werden.
Ich hoffe meine Antwort hilft Ihnen weiter. Ansonsten haken Sie gerne nach.
Herzliche Grüße
Nils Becker
Hallo Herr Dr. Becker,
wir handeln mit Medizinprodukten und sind dabei auf folgende Fragestellung gestoßen:
Ein Hersteller bringt das „Gebrauchsanweisung beachten“ Symbol aus der ISO 15223-1 auf das Label seines Medizinproduktes auf, legt allerdings den Produkten keine IFU bei.
Nach MDR Artikel 14 müssen wir als Händler prüfen, ob die notwendigen Informationen (inkl. IFU) dem Produkt beiliegen, was hier nicht der Fall ist.
Auf Nachfrage verweist der Hersteller auf die elektronsiche Gebrauchsanweisung, die auf der Webseite verfügbar ist. Allerdings findet sich kein eIFU Symbol oder ein Verweis auf die entsprechende Webseite auf dem Produktlabel. Der Hersteller verweist außerdem auf die im Artikel oben zitierte Ausnahme in MDR Anhang I, 23.1 d), da es sich um ein Klasse IIa Produkt handelt. Bringt der Hersteller aber nicht mit dem Aufbringen des Symboles zum Ausdruck, dass eine sichere Verwendung des Produkts nur mit IFU gewährleistet ist?
Oder wäre das nur der Fall wenn sich das andere von Herrn Schall erwähnte Symbol ISO 7010-M002 auf dem Label befindet?
Das würde aber bedeuten, das ein normales Gebrauchtanweisungsymbol auf einem Klasse I / IIa Produkt nicht bedeutet, dass die Gebrauchsanweisung dem Produkt beiliegen muss, was für meine Begriffe wenig Sinn ergibt.
Beste Grüße
Benedikt Brenk
Lieber Herr Brenk,
vielen Dank für die spannende Frage.
Es stimmt, für Klasse IIa Produkte muss nicht zwangsläufig eine IFU bereitgestellt werden. Allerdings nur unter der Bedingung, dass das Produkt auch ohne IFU sicher und effektiv benutzt werden kann. Das muss der Hersteller im Rahmen des Risikomanagements und Usability Engineerings nachweisen können. Ist in der Risikoanalyse das Bereitstellen einer IFU als Risikokontrollmaßnahme festgelegt, muss die IFU bereitgestellt werden und das Produkt bzw. die Verpackung mit dem Symbol ISO 7010-M002 entsprechend gelabelt werden.
Eine eIFU ist nur für ganz bestimmte Produkttypen erlaubt. Sehen Sie dazu bitte unseren Artikel zu eIFUs: https://www.johner-institut.de/blog/regulatory-affairs/elektronische-gebrauchsanweisung-medizinprodukte-207-2012/ . Wenn das Produkt nicht die entsprechenden Voraussetzungen für eine eIFU erfüllt, muss die IFU in Papierform dem Produkt beiliegen. Dies gilt laut MDR immer, sobald es eine IFU gibt, also auch für Klasse IIa Produkte, bei denen die IFU keine Risikokontrollmaßnahme darstellt.
Falls eine eIFU erlaubt und vorhanden ist, muss auf dem Labeling (Produkt, Verpackung oder beiliegendem Zettel, etc.) darauf aufmerksam gemacht werden und wie und wo auf diese zugegriffen werden kann (siehe Artikel 6 der VO 2021/2226).
Ich hoffe meine Antwort hilft Ihnen weiter.
Herzliche Grüße
Nils Becker
Hallo Herr Becker,
herzlichen Dank für Ihre schnelle und hilfreiche Antwort.
Dann scheine ich mit meiner Vermutung, dass die Argumentation des Herstellers nicht ganz schlüssig ist, wohl richtig zu liegen.
Wie vieles ist es aber mal wieder relativ schwierig, dass aus der MDR heraus einfach zu verstehen. Deshalb nochmals herzlichen Dank für Ihre Analyse.
Beste Grüße
Benedikt Brenk
Liebes Johner-Team,
können Sie aus Ihrem reichen Erfahrungsschatz berichten, wie mit Video-Aufnahmen umzugehen ist, die als Gebrauchsanweisung/Schulungsmaterialien (Tutorials) für Medizinprodukte bereitgestellt werden?
Gibt es hierzu Regelungen/Empfehlungen, z.B. zur Lenkung innerhalb der Technischen Dokumentation oder zur Validierung im Rahmen einer summativen Evaluation?
Besten Dank vorab und freundliche Grüße
Sonja Oechsler
Liebe Frau Oechsler,
vielen Dank für Ihre spannende Frage.
Videos sind per Definition keine Gebrauchsanweisung und dürfen diese auch nicht ersetzen. Dennoch können Videos als Teil der „Begleitdokumentation“ eine wertvolle Ergänzung zu der Gebrauchsanweisung sein und/oder für Schulungszwecke verwendet werden. Die Videos wären als User Interface zu sehen und unterlägen somit dem Usability-Engineering-Prozess nach IEC 62366-1. Das bedeutet, dass die sichere Gebrauchstauglichkeit der Videos nachgewiesen werden sollte, ggf. im Rahmen von summativen Evaluierungen. Somit wären die Videos auch Teil Ihrer Technischen Dokumentation und unterliegen Ihrem Dokumentenlenkungsprozess.
Ich hoffe meine Antwort hilft Ihnen weiter, ansonsten haken Sie gerne nach.
Herzliche Grüße
Nils Becker
Liebes Johner-Team,
wir haben in unseren Gebrauchsanweisungen eine Tabelle zur Materialinformation, wo ganz high level dargestellt wird, welcher Teil des Medizinprodukts aus welchem Material besteht. Wir haben uns im Team gefragt, ob diese Angabe überhaupt zwingend notwendig ist und aus welcher Norm diese Anforderung stammen könnte? Wir können es nicht mehr nachvollziehen.
Vielen Dank im Voraus und viele Grüße,
L. Hillerbrand
Sehr geehrte Frau Dr. Hillenbrand,
vielen Dank für Ihre spannende Frage.
Auch uns sind keine entsprechenden regulatorischen Anforderung bekannt, die besagen, man müsse in der IFU zwingend die Materialinformationen des Medizinprodukts angeben. Falls das Produkt allerdings CMR-Stoffe oder endokrin wirkende Stoffe mit einem Massenanteil größer 0,1 % enthält, müssen diese Stoffe angegeben werden (siehe MDR, Anhang I, 10.4.1, 10.4.5 und 23.4.a).
Ansonsten könnte die Angabe der Materialien sinnvoll sein, wenn man z.B. bei einer Wiederaufbereitung eine andere Prozesschemie verwenden will. Dies ist aber wie gesagt nicht regulatorisch gefordert.
Ich hoffe das beantwortet Ihre Frage, ansonsten haken Sie gerne nach.
Herzliche Grüße
Nils Becker
Liebes Johner-Team,
beziehen sich die Anforderungen, IFU, Artikelbezeichnung und Konformitätserklärung in Landessprache zur Verfügung zu stellen bereits auf Legacy Produkte, oder erst auf Produkte, für welche die Konformitätserklärung gem. (EU) 2017/745 ausgestellt wurde?
Vielen Dank,
S. Beck
Lieber Herr Beck,
vielen Dank für Ihre spannende Frage. Prinzipiell gelten für die Kennzeichnung von Legacy Produkte weiterhin die Anforderungen der Richtlinie 93/42/EWG (MDD). Die Anforderungen, dass die IFU, Artikelbezeichnungen und Konformitätserklärungen in Landessprache zur Verfügung zu stellen sind geht jedoch auf nationalen Anforderungen unabhängig von der MDR zurück und demzufolge gelten diese Anforderungen bereits unter der Richtlinie 93/42/EWG (MDD) und somit auch für Legacy Produkte.
Liebe Grüße
Philipp Schleer
Hallo 🙂
Können wir eine Gebrauchsanweisungen für mehrere Medical Devices (versch. inteded uses) erstellen? oder muss pro Intended use ( MD – Gruppe) ein separates IFU erstellt werden?
Liebe Grüsse
Sehr geehrte Frau Kabak,
aus regulatorischer Sicht spricht nichts dagegen, für mehrere Produkte nur eine Gebrauchsanweisung (ein Dokument) zur Verfügung zu stellen. Wichtig ist nur, dass für jedes Produkt spezifisch alle regulatorisch geforderten Angaben in der Gebrauchsanweisung gemacht werden. Aus ergonomischer Sicht sollte man bedenken, dass die vorgesehen Anwender die Gebrauchsanweisung leicht navigieren und problemlos die richtigen Abschnitte dem richtigen Produkt zuordnen können sollten . Dementsprechend kann es empfehlenswert sein, eher separate Gebrauchsanweisungen (Dokumente) zur Verfügung zu stellen.
Ich hoffe meine Antwort hilft Ihnen weiter, ansonsten haken Sie gerne nach.
Herzliche Grüße
Nils Becker
Hallo Hr. Becker,
besteht nun ein vom NB einforderbarer Zwang die IFU (oder ein Inhaltlich nach GSPR Anhang I 231. vollständiges alternatives Dokument) auf die Webseite hoch zu laden? Sie machen berechtigt den Hinweis auf die dann dann geltenden Forderungen.
Für unsere Produkte macht es einfach keinen Sinn, da jedes einzelne Produkt mit der jeweils gültigen IFU ausgeliefert wird zusammen in einer Faltschachtel. Die Produkte werden ausschließlich von Professionell Usern verwendet.
viele Grüße
Markus Manger
Lieber Herr Manger,
vielen Dank für Ihre Nachfrage. Die MDR fordert eindeutig, dass die IFU bzw. die geforderten Angaben des Anhang I Kapitel 23 auf der Webseite des Herstellers zur Verfügung gestellt werden muss, sofern der Hersteller eine Webseite hat. Im Englischen heißt es im Wortlaut: „Such information may appear on the device itself, on the packaging or in the instructions for use, and shall, if the manufacturer has a website, be made available and kept up to date on the website, taking into account the following: […]“.
Über die Sinnhaftigkeit dieser Anforderung mag man diskutieren, aber sie besteht und somit müssen die Benannten Stellen auch die Einhaltung überprüfen.
Ich hoffe meine Antwort hilft Ihnen weiter.
Herzliche Grüße
Nils Becker
Sehr geehrter Herr Becker,
Bestehen Checklisten, die es ermöglichen, einen bereits existierenden Leitfaden zu evaluieren? Dies wäre hilfreich, um Schwachstellen zu identifizieren und zu verbessern. Hierbei ist nicht nur der inhaltliche Aspekt von Bedeutung, sondern auch die Darstellung der Informationen.
Beste Grüsse
M. Baumann
Sehr geehrte Frau Baumann,
vielen Dank für Ihre Frage. Zu dem Thema benutzerfreundliche Gestaltung von Gebrauchsanweisung finden sich im Internet sicher zahlreiche kostenlose Guides und Checklisten, ohne dass ich persönlich eine besonders hervorheben könnte.
Ich kann allerdings die IEC/IEEE 82079-1 empfehlen. Diese Norm hat zwar keinen Branchenfokus, gilt aber auch für Medizinprodukte als Stand der Technik wenn es um Inhalt, Struktur und Form (Darstellung) der Gebrauchsanweisung geht.
Ich hoffe meine Antwort hilft Ihnen weiter. Falls nicht, haken sie gerne nach.
Herzliche Grüße
Nils
Hallo liebes Johner Team,
wir planen eine mehrsprachige GA für unterschiedliche Zielmärkte zu erstellen (z.B. EU, USA und Brasilien). Wenn es Anforderungen an den Inhalt der GA gibt, die nur für einen bestimmten Zielmarkt gelten, spricht in dem Fall etwas dagegen diese speziellen Anforderungen nur in dem Teil der GA mit der für den Zielmarkt geltenden Amtssprache aufzunehmen? Oder ist ihnen eine Anforderung bekannt, die besagt, dass alle Informationen in der IFU (in allen Sprachen) identisch sein müssen, so dass man bereits in der „Master-GA“ (i.S.v. Grundlage für die Übersetzungen der GA) eine Differenzierung von speziellen Zielmarkt-Anforderungen einarbeiten muss?
Herzlichen Dank im Voraus und viele Grüße
Peter
Lieber Herr Koszolka,
herzlichen Dank für Ihre Frage!
Mir ist keine regulatorische Anforderung bekannt, dass die IFU für die verschiedenen regulatorischen Zielmärkte inhaltlich identisch sein muss. Wie sie richtig schreiben, muss aber gewährleistet werden, dass die IFU die jeweiligen spezifischen Anforderungen des Markts erfüllt.
Ich hoffe ich konnte Ihnen weiterhelfen. Ansonsten fragen Sie gerne nach.
Herzliche Grüße
Nils Becker
Sehr geehrte Damen und Herren,
ist es legal sich via Gebrauchsanweisung selbst in ein aktives (nicht Anlage I) MP-Produkt einzuweisen und auf Grundlage dessen weiteres Personal einzuweisen? Bisher habe ich kein Widerspruch zur MPBetreibV gefunden, da dort nur für aktive Anlage I-Geräte eine Einweisung durch Hersteller bzw. beauftragte Person vorgesehen ist. Für aktive MP, welche nicht zur Anlage I gehören, muss lediglich eine Dokumentation der Einweisung erfolgen. Wer das macht und welche Einweisungs-Voraussetzungen der Einweisende mitbringt ist nicht geregelt.
Liege ich mit dieser Annahme richtig?
Vorab vielen Dank,
VG
Lieber Herr Gäbler,
leider ist das in der MPBetreibV für Medizinprodukte, die nicht unter die Anlage 1 fallen, nicht so eindeutig geregelt. Das BMG hat jedoch dazu eine Empfehlung auf der Website veröffentlicht: https://www.bundesgesundheitsministerium.de/faq-mpbetreibv
Dort finden Sie unter § 4 bei der 4. Frage „Welche Vorgaben sind bei der Einweisung zu beachten?“ folgende Antwort:
„§ 4 MPBetreibV enthält keine Vorgaben zur Art der Einweisung. Folglich wäre auch eine Einweisung durch Filme oder Software grundsätzlich zulässig. Es sollte aber einfache Möglichkeiten zu Rückfragen und Korrekturen der einzuweisenden Personen geben, z.B. eine kostenlose Telefonnummer. Es genügt demnach nicht, dass der Hersteller nur ein Anwendungs-Video zur Verfügung stellt, sondern setzt die Anwesenheit oder Erreichbarkeit einer einweisenden Person voraus. Auch in Bezug auf die einweisende Person gibt es keine Vorgaben in § 4. Es wird aber empfohlen, sich hier nach den Regelungen des § 10 für Medizinprodukte der Anlage 1 zu richten. Das heißt, dass die einweisende Person vom Hersteller oder einer Person, die im Einvernehmen mit dem Hersteller handelt, eingewiesen wurde, bevor sie die Einweisung anderer Anwenderinnen und Anwender übernimmt. Entscheidend ist, dass eine entsprechend kompetente Person in geeigneter Weise in die sachgerechte Handhabung einweist.“
Aufgrund dessen würde ich von der Selbsteinweisung anhand der Gebrauchsanweisung abraten und wie beschrieben auch bei nicht Anlage 1 Produkten die Einweisung durch den Hersteller, eine andere durch den Hersteller dazu beauftragte Person oder die durch diese beiden Personen eingewiesene beauftragte Person des Betreibers durchführen lassen.
Herzliche Grüße
Christopher Seib
Sehr geehrter Herr Seib,
vielen Dank für die ausführliche Antwort und die Empfehlung!
Viele Grüße
Sehr geehrte Damen und Herren,
ich wollte fragen, ob die MDR zwischen einer Gebrauchsanweisung für Klasse I-Produkte vs. Klasse IIa-Produkte unterscheidet. Sind hier die Anforderungen gleich, sodass beide IFUs das gleiche beinhalten müssen?
Vielen Dank im Voraus und freundliche Grüße
Nam Nguyen
Sehr geehrter Herr Nguyen,
die MDR macht an sich keine Unterscheidungen bei den Inhalten der IFU zwischen den unterschiedlichen Klassen (außer Punkte wie der SSCP, welche per Definition nicht für niedrige Risikoklassen erforderlich ist). Je höher die Klasse, desto höher auch das Risiko – daher sind bei einer Klasse IIa-Anleitung vermutlich mehr Warnungen und ähnliches enthalten, aber rein regulatorisch sind die Vorgaben für beide identisch.
Herzliche Grüße
Christopher Seib
Liebes Johner-Team,
wir stellen ein Medizinprodukt her, für das nach derzeitigem Stand keine rein elektronischen Gebrauchsanweisungen erlaubt sind. Wir verkaufen ausschließlich B2B, jedoch Produkte, die schlussendlich von Endverwendern gekauft und genutzt werden. Es kam nun die Frage auf, ob es möglich ist, unseren Vertriebspartnern in den unterschiedlichen EU-Ländern, die Gebrauchsanweisung ausschließlich elektronisch/per pdf zur Verfügung zu stellen, sodass diese sie selbst ausdrucken und dann in Papierform an ihre Kunden (Händler und / oder Endverwender) weitergeben können. Um sicherzustellen, dass immer die aktuelle Version in Papierform an deren Kunden gegeben wird, wäre es nötig/ausreichend, hier eine schriftliche Vereinbarung mit den einzelnen Vertriebspartnern zu schließen?
Oder müssen zwingend wir als Hersteller die Gebrauchsanweisungen in Papierform bereits mit den gelieferten Medizinprodukten an unsere Vertriebspartner in den unterschiedlichen EU-Ländern liefern?
Vielen Dank für Ihre Expertise/Einschätzung!
Liebe Martha,
ohne eine beiliegende Gebrauchsanweisung „fehlt“ dem Produkt noch etwas, um die MDR (hier Artikel 10(11)) komplett zu erfüllen. Daher wäre das Beifügen der korrekten, vom Hersteller freigegebenen Version und das Weiterleiten mit jedem Produkt an den Anwender ein ausgelagerter Prozess. Für diesen Prozess benötigt Ihr Partner eine Verfahrensanweisung und muss Artikel 16(3) der MDR einhalten.
Entsprechend sollten Sie neben einem Vertrag, der die Arbeiten Ihres Partners abdeckt, auch die Güte des Prozesses sicherstellen, zum Beispiel über interne Audits. Dann ist das Beifügen über den Vertriebspartner für mein Verständnis möglich.
Herzliche Grüße
Christopher Seib
Guten Tag,
eigentlich würden wir für unsere „IVDs für den professionellen Gebrauch“ vornehmlich englischsprachige IFUs anbieten.
Die Regelung nach §8 (2) MPDG lässt dies im ersten Halbsatz auch zu, allerdings sind sicherheitsrelevante Informationen IMMER auch Bestandteil einer IFU, so dass dieser Teil in DE nur in deutscher Sprache formuliert werden darf. Meine objektive Auslegung des MPDG ist aber in diesem Fall die, dass man es den Herstellern mit dieser Regelung einfacher machen wollte, da die professionellen Anwender durchaus der englischen Sprache mächtig sein müssen.
Wie sehen sie das?
Sehr geehrter Herr Breß,
das MPDG ist hier leider etwas schwammig: “ in deutscher Sprache oder in der Sprache des Anwenders“. Die Sprache des Anwenders würde ich bei professionellen Anwendern in Ihrem Fall als Englisch auslegen, da dies generell verständlich sein sollte – ganz ähnlich sieht das auch die EU (https://health.ec.europa.eu/publications/overview-language-requirements-manufacturers-medical-devices_en).
Beachten Sie aber auch, dass Sie möglicherweise andere Verordnungen (zum Beispiel an das Sicherheitsdatenblatt nach EG 1907/2006: „Das Sicherheitsdatenblatt wird in einer Amtssprache des Mitgliedstaates/der Mitgliedstaaten vorgelegt, in dem der Stoff oder die Zubereitung in Verkehr gebracht wird, es sei denn, der betreffende Mitgliedstaat bestimmt/die betreffenden Mitgliedstaaten bestimmen etwas anderes.“) einhalten müssen.
Herzliche Grüße
Christopher Seib