
Die Medizinprodukteverordnung hat die Anforderungen an die Händler deutlich erhöht. Lernen Sie diese Anforderungen zu verstehen, um die mehrjährigen Freiheitsstrafen zu vermeiden, die bei einem Verstoß drohen.
Dieser Beitrag berücksichtigt auch eine umfangreiche Leitlinie der irischen Aufsichtsbehörde.
1. Händler: Definition & Abgrenzung
a) Definition
Die Medizinprodukteverordnung (MDR) hat viele Rollen eingeführt, darunter die der Wirtschaftsakteure. Zu diesen Wirtschaftsakteuren zählen neben den Herstellern, den EU-Bevollmächtigten und den Importeuren auch die Händler.
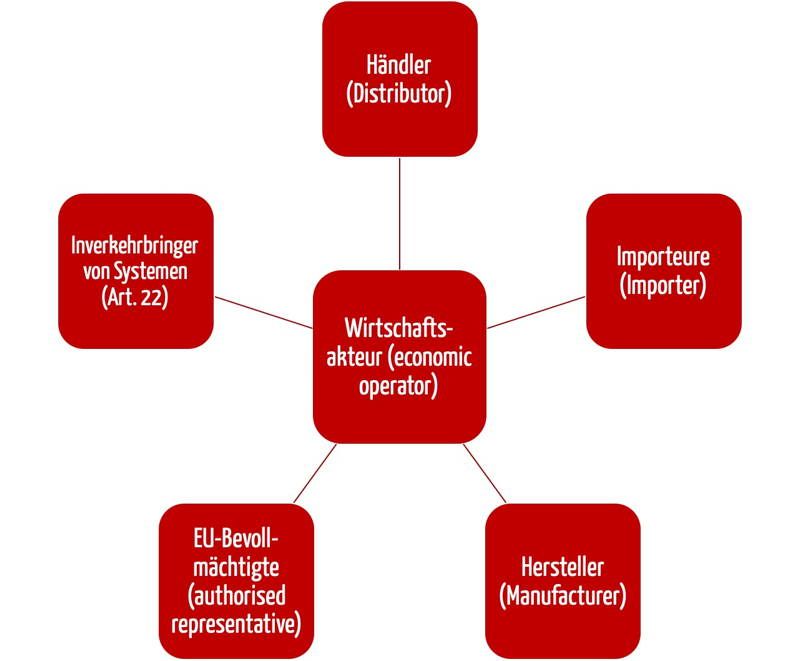
„Händler“ bezeichnet jede natürliche oder juristische Person in der Lieferkette, die ein Produkt bis zum Zeitpunkt der Inbetriebnahme auf dem Markt bereitstellt, mit Ausnahme des Herstellers oder des Importeurs
Quelle: MDR, Artikel 2
Die Händler sind somit die Personen oder Organisationen, die Medizinprodukte verkaufen, sei es an Endverbraucher oder an Zwischenhändler.
b) Typische Tätigkeiten beim Vertrieb von Medizinprodukten
Zu den typischen Tätigkeiten eines Händlers zählen:
- Kauf der Medizinprodukte vom Hersteller oder anderen (Zwischen-)Händlern
- Marketing und Verkauf von Medizinprodukten an Endkunden oder andere Händler
- Lagerung und Transport von Produkten
- Ggf. Aufbringen eigener Labels (Vorsicht damit! Dazu später mehr.)
- Einweisung von Anwendern
- Unterstützung bei der Installation und Inbetriebnahme
- Beantworten von Fragen der Anwender
- Behandeln von Kundenbeschwerden, Rückmeldung an Hersteller
Manche Händler übernehmen auch die Wartung und Reparatur der Produkte oder organisieren dies.
c) Abgrenzung der Rollen Händler und Importeur
Wenn ein Händler Produkte von einem Hersteller oder einem anderen Händler einkauft, der nicht in der EU angesiedelt ist, übernimmt dieser Händler zusätzlich die Rolle des Importeurs. Die MDR definiert Importeure wie folgt:
„Importeur“ bezeichnet jede in der Union niedergelassene natürliche oder juristische Person, die ein Produkt aus einem Drittland auf dem Unionsmarkt in Verkehr bringt
Quelle: MDR, Artikel 2
An Importeure stellt die MDR zusätzliche Anforderungen.
d) Abgrenzung der Rollen Händler und Hersteller
Viele Händler möchten unter eigenem Namen die Medizinprodukte verkaufen und den eigentlichen Hersteller nicht nennen. Damit wird der Händler zum Hersteller, denn die Medizinprodukteverordnung definiert Hersteller so:
„Hersteller“ bezeichnet eine natürliche oder juristische Person, die ein Produkt herstellt oder als neu aufbereitet bzw. entwickeln, herstellen oder als neu aufbereiten lässt und dieses Produkt unter ihrem eigenen Namen oder ihrer eigenen Marke vermarktet
Quelle: MDR, Artikel 2
Mit der MDR gehören auch die PLM-OEM-Konstrukte der Vergangenheit an. Dafür bietet die MDR gemäß Artikel 16, Absatz 2 den Händlern neue Möglichkeiten. Sie schätzt folgende Tätigkeiten nicht mehr als eine Produktänderung ein, die Auswirkung auf die Konformität des Produkts hat:
- Bereitstellung von Informationen für ein Produkt. Das gilt auch für Übersetzungen.
- Änderung der äußeren Verpackung
Natürlich wäre es auch denkbar, dass der Hersteller das Produkt im „Design“ des Händlers in den Verkehr bringt. Der Hersteller muss aber immer (auch) auf dem Label erkennbar sein. Das fordert Artikel 16, Absatz 3.
2. Pflichten der Händler
Bereits bevor die Händler die Medizinprodukte verkaufen, müssen sie viele gesetzliche Anforderungen erfüllen.
a) Hintergrund und Historie
Die Anforderungen, die die Medizinprodukteverordnung MDR an die Händler stellt, leiten sich aus einem übergeordneten Rahmenwerk zur Vermarktung von Produkten ab. Dieses ist auch als „Goods Package“ bekannt und geht zurück auf:
- EU-Verordnung 765/2008 „über die Vorschriften für die Akkreditierung und Marktüberwachung im Zusammenhang mit der Vermarktung von Produkten“
- Beschluss Nr. 768/2008/EG „über einen gemeinsamen Rechtsrahmen für die Vermarktung von Produkten“
b) Pflichten der Händler als Prüfinstanz in der Lieferkette (aus Sicht der MDR)
Der Gedanke der MDR besteht darin, dass jeder Wirtschaftsakteur nach Möglichkeit sicherstellt, dass der Wirtschaftsakteur einen Schritt zuvor in der Lieferkette die regulatorischen Anforderungen erfüllt hat.

Zu diesen Überprüfungen, die die MDR von den Händlern verlangt, zählen:
- Trägt das Produkt eine CE-Kennzeichnung?
- Wurde für das Produkt eine Konformitätserklärung ausgestellt?
- Hat der Importeur seinen Namen und seine Anschrift auf dem Produkt, der Verpackung oder einem Dokument angegeben?
- Hat der Importeur mit seinen zusätzlichen(!) Kennzeichnungen nicht die Kennzeichnungen des Herstellers verdeckt?
- Hat der Hersteller die UDI vergeben?
- Scheint das Produkt konform mit den gesetzlichen Vorgaben zu sein?
Wenn eine dieser Bedingungen nicht erfüllt ist, darf der Händler das Produkt nicht bereitstellen und muss ggf. den Hersteller, den Importeur und den EU-Repräsentanten informieren.
Händler dürfen sich damit nicht mehr blind auf die Hersteller und Importeure verlassen. Sie sollten insbesondere in der Übergangszeit genau überprüfen, ob die Hersteller gültige Zertifikate vorlegen.
c) Pflichten der Händler als Prüfinstanz in der Lieferkette (aus Sicht des MPDG)
Anforderungen des MPDG
Die MDR erlaubt den Händlern in Artikel 14 Absatz 2, Stichprobenprüfungen durchzuführen. Sie sollen damit überprüfen können, ob die Produkte tatsächlich eine CE-Kennzeichnung, eine Konformitätserklärung und eine UDI haben.
Das Medizinproduktedurchführungsgesetz MPDG erscheint rigider: Es droht in § 92 Abs. 1 Nr. 3 in Verbindung mit § 13 MPDG mit einer Freiheitsstrafe, wenn jemand gefälschte Produkt anbietet, auf Lager hält oder in Betrieb nimmt. Das betrifft die Händler.
Das Verbot in § 13 MPDG und die dazugehörige Strafandrohung ist im Zusammenhang mit Art. 14 Abs. 2 Unterabsatz 3 MDR zu sehen: Danach bestehen ein Vertriebsstopp und eine zusätzliche Meldepflicht an die zuständige Behörde, wenn ein Händler „der Auffassung ist oder Grund zur Annahme“ hat, dass ein Produkt gefälscht ist und damit nicht konform.
Der Händler hat unserer Einschätzung nach jedoch weder hieraus noch aus § 92 Abs. 1 Nr. 3 und § 13 MPDG die regulatorische Pflicht, über die stichprobenartigen Prüfungen hinaus auf etwaige Fälschungen zu prüfen, d. h. per Default misstrauisch zu sein und darauf basierend eine 100%-Prüfung festzulegen. Für die Strafvorschrift kommt es unserer Einschätzung nach auch darauf an, ob das unzulässige Anbieten oder Lagerhalten tatsächlich gefälschter Ware fahrlässig oder gar vorsätzlich erfolgte. Es wird somit kritisch, wenn der Händler weitere Anhaltspunkte oder Verdachtsmomente aufzeigt, dass er Fälschungen vertreibt.
Sich daraus ergebende mögliche Konflikte
Wenn der Händler ein gefälschtes Produkt entdeckt und im Lager behält, macht er sich strafbar. Nach § 13 MPDG ist jedoch auch das Zurücksenden fraglich, denn dann bringt er es ggf. „aus dem Geltungsbereich dieses Gesetzes“. Das ist auch strafbar.
Dann bleibt noch die Möglichkeit, es zu vernichten. Doch auch in diesem Fall macht er sich vermeintlich strafbar, weil er den Behörden „auf Ersuchen unentgeltliche Proben des Produkts zur Verfügung stellen“ oder, „sofern dies nicht praktikabel ist, Zugang zu dem Produkt verschaffen muss“ (MDR Artikel 14(6)).
Ein Leser, der auf diesen Konflikt aufmerksam machte, schreibt:
„Da bin ich mal auf die Handreichung des Gesetzgebers gespannt, wie diese Widersprüche von einem Händler gelöst werden sollen, z.B. vom Aldi, der ja auch ein- bis zweimal im Jahr Medizinprodukte (z. B. Blutdruckmessgeräte) im Rahmen irgendwelcher Aktionen führt.“
d) Anforderungen an die Tätigkeiten der Händler
Die MDR verpflichtet die Händler zu weiteren Tätigkeiten:
- Produkte nur gemäß den Vorgaben des Herstellers lagern und transportieren
- Beschwerden sowie Berichte über Vorkommnisse sammeln und an die Hersteller und ggf. Importeure weiterleiten. Das gilt auch für Produkte, von denen die Händler selbst Zweifel an der Konformität haben.
- Ein „Register“ nichtkonformer Produkte, Rückrufe und Rücknahmen führen
- Behörden (in Deutschland BfArM gemäß § 44 MPDG) über unsichere und gefälschte Produkte sowie über Korrekturmaßnahmen informieren und diesen auf Verlangen Informationen und Unterlagen aushändigen
Mit der MDR werden die Händler wie die Importeure Teil des Post-Market-Surveillance- und Meldesystems. Sie müssen aktiv daran mitwirken, was bedingt, dass sie die Produkte nachverfolgen können. Das schreibt die MDR explizit in Artikel 25 und die EU in ihrem Factsheet for Authorized Representatives, Importers and Distributors.
e) Registrierung von Händlern
Die MDR spricht in Artikel 30 zwar von dem „Elektronischen System für die Registrierung von Wirtschaftsakteuren“. Sie verpflichtet darin aber nur die Hersteller, Bevollmächtigten und Importeure. Die MDR überlässt es den Mitgliedsstaaten, Bestimmungen für die Registrierung von Händlern zu erlassen.
Genau das scheinen Mitgliedsstaaten wie Deutschland auch zu tun: Im Medizinproduktedurchführungsgesetz MPDG heißt es:
(1) Das Bundesministerium für Gesundheit wird ermächtigt, …
9. festzulegen, dass Händler, die Produkte auf dem deutschen Markt bereitstellen, dies vor Aufnahme ihrer Tätigkeit bei der zuständigen Behörde anzuzeigen haben, sowie Inhalt und Form der Anzeige zu regeln.
MPDG § 88
Damit ist eine nationale Verordnung zu erwarten, die diese Registrierungspflicht beschreiben wird.
f) Sonderfall Qualitätsmanagementsystem
Wann Händler ein Qualitätsmanagementsystem benötigen
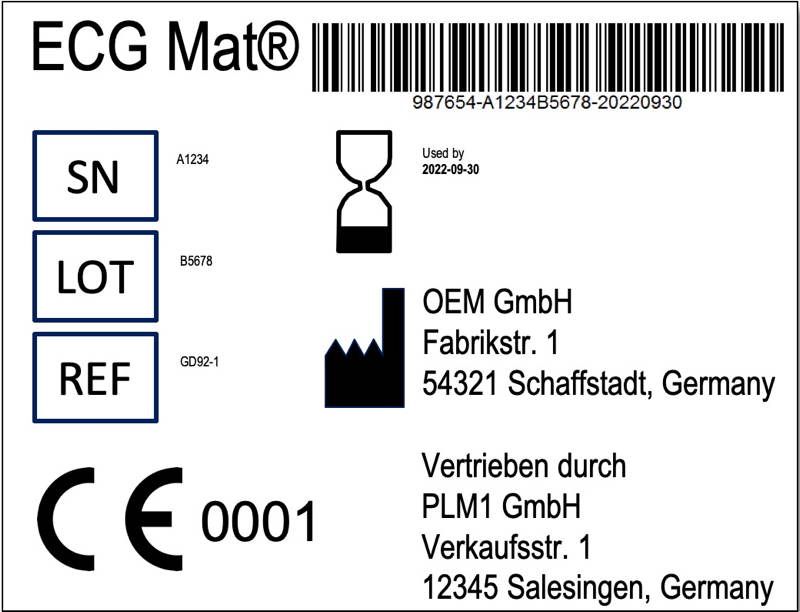
Wie oben beschrieben, dürfen Händler Produkte umverpacken und gesetzliche geforderte Informationen (Anhang I, Abschnitt 23) bereitstellen und übersetzen. Daraus ergibt sich aber nicht nur die bereits erwähnte Pflicht, den Namen des Händlers auf dem Label anzugeben (s. Abb. 2). Vielmehr muss der Händler über ein Qualitätsmanagementsystem verfügen.
Dieses QM-System muss alle relevanten Tätigkeiten regeln:
- Umverpacken des Produkts (und prüfen, dass dieses Umverpacken die Konformität nicht beeinträchtigt)
- Erstellen/Bereitstellen der Informationen (und prüfen, dass diese den gesetzlichen Anforderungen genügen)
- Übersetzen der Informationen
- Informationen über Korrekturmaßnahmen des Herstellers erhalten/einfordern(?)
Nach Aussage der irischen Behörde muss das QM-System nicht zertifiziert sein: “it is not a requirement that a quality system is officially accredited to any specific standard“. Das steht jedoch im Widerspruch zur Forderung der MDR
Within the same period of 28 days, the distributor or importer shall submit to the competent authority a certificate, issued by a notified body designated for the type of devices that are subject to activities mentioned in points (a) and (b) of paragraph 2, attesting that the quality management system of the distributer or importer complies with the requirements laid down in paragraph 3.
MDR Artikel 16, Absatz 4
Welche Prozesse ein Qualitätsmanagementsystem festlegen sollte
Ob dieses QM-System einen bestimmten Standard (z. B. einer Norm) erfüllen und nach einem bestimmten Standard geprüft werden muss, erscheint unklar. Nach Ansicht der irischen Behörde muss dieses QM-System relativ umfassend sein und auch weitere Tätigkeiten lenken, z. B.:
- Personal, Training
- Lenkung von Dokumenten und Aufzeichnungen
- Empfang, Lagerung und Bereitstellung von Medizinprodukten
- Umgang mit zurückgegebenen Produkten
- Umgang mit gefälschten Produkten
- Rückruf von Produkten
- Ausgelagerte Prozesse
- Transport
- Audits
- Lieferantenlenkung
- Management Review
- CAPA
- Abfallmanagement
- Interne und externe Audits
- Validierung inkl. Computerized Systems Validation
Beachten Sie als
- Händler: Gemäß der Behörde müssen Sie ein vollständiges QM-System vergleichbar ISO 13485 implementieren.
- Händler: Sie müssen im Fall von Artikel 16(4) der Behörde bescheinigen, dass ihr QM-System den Anforderungen ebendieses Artikels genügt.
In einer früheren Version des Artikels hieß es, dass der Hersteller die Bescheinigung vorlegen müsse. Dies ist nicht zutreffend. Es ist der Händler. Die deutsche Übersetzung ist leider falsch. Sie schreibt „Der Hersteller oder Importeur legt der zuständigen Behörde im selben Zeitraum von 28 Tagen eine Bescheinigung vor.“ Im englischen Text heißt es: „distributors or importers carrying […] shall inform the manufacturer and the competent authority of the Member State.“ Zudem wurde die Diskussion ergänzt, ob und wenn ja nach welchem Standard ein QM-System zu errichten und zu prüfen sei.
Wahrscheinlich damit die Behörde das QM-System prüfen kann, verpflichtet die MDR den Händler, sie 28 Tage vor der Bereitstellung des Produkts zu unterrichten. Die MDR spricht von „unterrichten“, nicht von „um Genehmigung bitten“.
Wir unterstützen Hersteller ebenso wie Händler dabei, schnell ISO 13485-konforme QM-Systeme zu etablieren und damit die gesetzlichen Auflagen zu erfüllen.
Kontakt aufnehmen
g) Pflichten, die die MDR den Händlern nicht(?) auferlegt
Die Händler sind nicht verpflichtet, die Produkte zu registrieren. Das ist die Aufgabe der Hersteller bzw. Importeure. Die Händler benötigen auch keine „Person Responsible for Regulatory Compliance“.
Unklar ist, ob der Händler für die Lagerung und den Transport verantwortlich ist, wenn der Hersteller direkt an die Kunden des Händlers liefert. Zu Diskussionen führt auch die Frage, ob der Importeur oder der Händler für den Transport vom Importeur zum Händler verantwortlich zeichnet.
h) Übergangsfristen
Händler betrifft v. a. die sogenannte Abverkaufsregelung der MDR Artikel 120, Absatz 4. Diese soll den „Zeitraum befristen, in dem AIMDD/MDD konforme Produkte, die bereits (erstmalig) in Verkehr gebracht wurden (entweder vor GB oder mittels Art. 120 Abs. 3 MDR nach GB), z. B. durch einen Händler bereitgestellt werden dürfen.“
Nach dem 27. Mai 2025 dürfen diese Produkte nicht mehr bereitgestellt/in Betrieb genommen werden (= Endtermin). Derartige Produkte, die sich an diesem Tag noch immer in der Handelskette befinden, – d. h. noch nicht dem Endanwender (z. B. Krankenhaus) als gebrauchsfertiges Produkt zur Verfügung gestellt wurden – sind nicht mehr „handelbar“.
NAKI UG1
Art. 120 Abs. 4 MDR adressiert im Wesentlichen das „Bereitstellen“ auf dem Markt von AIMDD/MDD-konformen Produkten, nachdem sie (erstmalig) in Verkehr gebracht wurden, z. B. in der Handelskette. Er regelt nicht das “(erstmalige) Inverkehrbringen” dieser Produkte durch den Hersteller.
Weiter stellt der NAKI klar, dass „der Handel mit Second-Hand-Produkten nicht von der sog. „Abverkaufsregelung“ erfasst sein soll (siehe Erwägungsgrund 32). Dies bedeutet, dass sobald ein Produkt dem Endanwender (z. B. Krankenhaus) als gebrauchsfertiges Produkt zur Verfügung gestellt wurde, das weitere Bereitstellen dieses Produktes auf dem Markt nicht dem Anwendungsbereich der MDR unterfällt.“
Die MDCG stellt in MDCG 2021-25 klar, dass die Wirtschaftsakteure ihren Pflichten nach MDR auch für die Legacy-Devices (Artikel 120, gültiges MDD-Zertifikat) nachkommen müssen. Die MDCG listet für jeden Wirtschaftsakteur die entsprechenden Artikel der MDR auf, die er beachten muss. Bei den Händlern sind das:
- Artikel 14(2) letzter Abschnitt: Hier geht es um die Informationspflichten im Rahmen der Post-Market Surveillance
- Artikel 14(4): Auch hier geht es um die Informationspflichten und das Mitwirken bei den Korrekturmaßnahmen
- Artikel 14(5): Auch hier geht es um das Mitwirken im Rahmen der Post-Market Surveillance und der damit verbundenen Maßnahmen.
- Artikel 14(6): Dieser Absatz betrifft die Kooperation der Hersteller mit den Behörden.
3. Hinweise für Hersteller (bzgl. Händler)
a) Problemstellung
Manche Hersteller mögen denken, dass all diese regulatorischen Vorgaben das Problem der Händler und nicht das eigene seien. Doch das wäre zu kurz gedacht:
Händler dürfen Produkte sogar „umkennzeichnen“ und „umverpacken“, z. B. im eigenen Design. Sie müssen die Hersteller nur darüber „unterrichten“, nicht um Genehmigung fragen.
Mit anderen Worten: Die Hersteller können einerseits diese Tätigkeiten nicht (einfach) unterbinden. Andererseits sind sie zur Post-Market Surveillance verpflichtet. Das wird schwierig, wenn Händler Produkte in weiteren, dem Hersteller nicht bekannten Ländern mit übersetzten Begleitmaterialien vertreiben.
b) Post-Market Surveillance
Hersteller sollten proaktiv und weltweit nach Informationen zu den eigenen Produkten suchen. Das betrifft explizit nicht die Länder, von denen sie wissen, dass ihre Produkte vertrieben werden. Hier hilft eine Automatisierung wie das Post-Market Radar des Johner Instituts.
Es ist unerlässlich, dass die Hersteller mit ihren Händlern, Bevollmächtigten und Importeuren genau regeln, wer für welche Tätigkeiten (nicht nur) im Rahmen der Post-Market Surveillance zuständig ist.
Dass diese Überwachung nach der Inverkehrbringung auch gefälschte Produkte und nicht autorisierte Händler im Fokus haben sollte, versteht sich von selbst.
c) Händler an die Kandare nehmen
Hersteller sollten die Händler genauso überwachen wie die Produkte selbst.
Händlern, die die Produkte „umverpacken“ oder „umkennzeichnen“, müssen der Behörde sogar ein Zertifikat einer Benannten Stelle vorlegen. Damit ist den Herstellern ein Teil der Arbeit abgenommen.
In einer früheren Version des Artikels hieß es, dass die Hersteller die Zertifikate vorlegen müssten. Dies ist allerdings ein Fehler in der deutschen Version der MDR. Dort hieß es „Der Hersteller oder Importeur legt…“, in der englischen Version heißt es „the distributor or importer shall submit …“
Dennoch können sich die Hersteller nicht blind darauf verlassen. Sie müssen ihre Händler kennen und überwachen.
Manchmal ist es hilfreich, den bestimmungsgemäßen Gebrauch und das Design der Produkte (z. B. Stecker) so zu wählen, dass bestimmte Märkte ausgeschlossen sind. Damit können Hersteller den „Missbrauch“ durch ungewünschte Händler etwas eindämmen.
Zudem sollten die Hersteller mit den Händlern Qualitätssicherungsvereinbarungen abschließen. So eine QSV sollte z. B. regeln:
- Betroffene Produkte bzw. Produktgruppen
- Geltungsdauer der Vereinbarung
- Ansprechpartner auf beiden Seiten
- Form und Fristen für Rückmeldungen der Händler an den Hersteller
- Recht des Herstellers auf Audit/Inspektion des Händlers
- Verpflichtung des Händlers zu Behördenmeldungen
- Verpflichtung des Händlers zum Nachverfolgen der Produkte („Register“)
- Ggf. Verbot zum Verkauf an andere Händler
- Verpflichtung des Händlers, Importeure und EU-Beauftrage zu informieren
- Bereithalten von Prüfmustern
- Verpflichtung, Begleitmaterialien bereitzulegen
- Verpflichtung, nur mit einer zertifizierten Übersetzungsagentur zu arbeiten
d) Vorsicht mit Fulfillment-Partnern
Bestimmte Logistikpartner bieten Dienste an, die über die eines üblichen Paketdienstleisters (z. B. DHL) hinausgehen. Diese sogenannten „Fulfillment Centers“ oder “Fulfillment Houses“ lagern Produkte, verpacken diese auf Wunsch, übernehmen die Fakturierung usw.
Dann spricht man nicht mehr von „neutralen Dienstleistern“, sondern von Händlern im Sinne der MDR.
Unser Tipp: Ziehen Sie die Vorgaben des Blue-Guides der EU zurate.
Die MDR hat die Anforderungen an die Händler substanziell erhöht. Das ist nachvollziehbar, weil nur alle an der Logistikkette Beteiligten gemeinsam nachvollziehen können, wo sich welche Produkte befinden und ob diese Produkte konform sind.
4. FAQ
a) Wer darf Medizinprodukte verkaufen?
Der Gesetzgeber schließt keine Personen oder Organisationen vom Verkauf von Medizinprodukten aus. Allerdings stellt der Gesetzgeber Anforderungen an diese Verkäufer und nennt diese Händler. Die Händlerpflichten wurden in diesem Artikel vorgestellt.
Medizinproduktehersteller können jedoch den Handel mit ihren Produkten (mit Einschränkungen) untersagen.
b) Welche Erlaubnis oder Berechtigung ist erforderlich, um mit Medizinprodukten handeln zu dürfen?
Es gibt keine offizielle Erlaubnis oder Berechtigung durch staatliche Stellen. Allerdings müssen die Händler die gesetzlichen Voraussetzungen erfüllen, bevor sie mit dem Handel beginnen dürfen. Zu diesen Voraussetzungen können zählen:
- Qualitätsmanagementsystem (für bestimmte Händler)
- Registrierung bei Behörden (z. B. in Deutschland; siehe oben)
- Prüfungen des Produkts (z. B. Angaben des Importeurs, UDI)
Falls ein Händler auch Importeur ist, kommen weitere Voraussetzungen hinzu, die er vor der Aufnahme des Handels erfüllen muss.
Zudem kann es sein, dass ein Hersteller Anforderungen an die Händler stellt. Es könnte etwa sein, dass der Händler einen Kompetenznachweis vorlegt oder beim Hersteller erbringt.
c) Wie bringt man ein Medizinprodukt auf den Markt?
Die EU unterscheidet genau zwischen der Inverkehrbringung, der erstmaligen Inverkehrbringung und der Bereitstellung. Händler sind die natürlichen oder juristischen Personen, die Produkte auf dem Markt bereitstellen.
Beachten Sie den Fachartikel zur Inverkehrbringung von Medizinprodukten, der auch die Abgrenzung zur Bereitstellung erklärt.
5. Fazit / Zusammenfassung
Nur gemeinsam können die Wirtschaftsakteure auf Nicht-Konformitäten reagieren und so die Sicherheit der Patientinnen und Patienten erhöhen. Damit nimmt ihnen die MDR die Möglichkeiten eines „Finger Pointings“.
Das sind wichtige Erkenntnisse:
- Die Händler sollten über ein (weitgehend) vollständiges Qualitätsmanagementsystem verfügen. Das ist zumindest die Sicht der irischen Behörde. Nicht nur für das Rückmeldesystem sollten Prozesse etabliert sein.
Für den Fall, dass ein Händler Tätigkeiten gemäß Artikel 16(2) durchführt, muss er über ein QM-System verfügen. Eine Pflicht zur Zertifizierung gibt es (derzeit) nicht. - Die Hersteller sollten sich bewusst sein, dass die Pflichten der Händler sie zumindest indirekt ebenfalls betreffen.
- Die irische Behörde hat eine Leitlinie für Händler mit weiteren Praxistipps veröffentlicht.
So bleibt zu hoffen, dass das MPDG nicht zu Widersprüchen mit der MDR führt und dass eine höhere Sicherheit der Patienten und nicht ein Mehr an Bürokratie das Ergebnis dieser gesetzlichen Verschärfungen sind.
- 2024-12-18: FAQ (Kapitel 4) eingefügt. Zwischenüberschriften bei 2.e) und 2.f) eingefügt. Hauptüberschriften gekürzt. Einige redaktionelle Verbesserungen.
- 2021-10-25: In Abschnitt 2.h) die Anforderungen durch die Leitlinie MDCG 2021-25 ergänzt

Sehr geehrter Herr Prof. Dr. Christian Johner,
vielen Dank für diesen sehr informativen Artikel. Eine Anmerkung habe ich jedoch. In Artikel 14, Abs. 5 der MDR heißt es:
„[…] Sie [(die Händler)] führen ein Register der Beschwerden, der nichtkonformen Produkte und der Rückrufe und Rücknahmen, und sie halten den Hersteller und gegebenenfalls dessen Bevollmächtigten und den Importeur über diese Überwachungsmaßnahme auf dem Laufenden und stellen ihnen auf deren Ersuchen alle Informationen zur Verfügung.“
oder im englischen:
„[…] They shall Keep a register of complaints, of non-conforming devices and of recalls and withdrawals, and keep the […]“
Sie haben den Complaints-Part unter „d) Anforderungen an die Tätigkeiten der Händler“ jedoch nur auf die Vorkommnisse bezogen. Ich würde die MDR jedoch an dieser Stelle so deuten, dass complaints über mutmaßliche Vorkommnisse, wie beschrieben, unverzüglich weitergeleitet werden müssen, aber auch complaints im Sinne der ISO 13485 in das Register mit aufgenommen werden müssen, zusätzlich zu den nichtkonformen Produkten, den Rückrufen und den Rücknahmen. Ich beziehe mich auf Ihre Punkte unmittelbar am Anfang von Abschnitt d).
Dieser Punkt ist aus meiner Sicht insofern wichtig, da er es Herstellern in Zukunft leichter macht, besser an Informationen der Händler über die eigenen Produkte zu kommen.
Ich würde mich über Ihre Meinung in dieser Angelegenheit freuen.
Sehr geehrter Herr Neustupny,
in meinem Text heißt es: „Beschwerden und Berichte über Vorkommnisse sammeln und an die Hersteller und ggf. Importeure weiterleiten.“ Es geht also explizit auch um die Beschwerden.
Ich habe das „und“ durch ein „sowie“ ersetzt, um klar zu machen, dass es zwei Punkte sind.
Ich hoffe, dass ich Ihre Frage richtig verstanden und adressiert habe.
Beste Grüße, Christian Johner
Danke für den erschreckenden Artikel. Meine Frage ist ob dies generell für alle Medizinprodukte gelten soll. Genauer sind auch Klasse 1 Produkte generell von diesen Vorgaben betroffen?
Sehr geehrte Frau Schunk,
ich wollte mit dem Artikel nicht erschrecken. Im Gegenteil: Ich bin sogar der Meinung, dass die Forderungen sinnvoll sind.
Die Anforderungen an die Händler unterscheiden sich nicht von der Klasse. Die Überwachung (durch benannte Stelle des Herstellers bzw. durch Behörde) unterscheidet sich aber.
Beste Grüße, Christian Johner
Hallo Herr Professor Johner,
Sie schreiben, dass eine Zertifizierung des QM-Systems von Händlern nicht notwendig ist. Im Gegensatz dazu steht im OEM-Artiekl, dass das QM-Systems eines Händlers (nicht PLM) durch eine Benannte Stelle begutachtet werden muss. Können Sie das aufklären?
Vielen Dank,
Kerstin
Sehr geehrte Kerstin,
die benannte Stelle des Herstellers hat das Recht, ausgelagerte Prozess zu auditieren — hier die des Händlers. D.h. aber nicht, dass das QM-System des Händlers auch zertifiziert werden muss. D.h. Abweichungen, die die benannte Stelle findet, betreffen zuerst das QM-System des Herstellers, weil dieser ausgelagerte Prozesse nicht ausreichend lenkt.
Beste Grüße, Christian Johner
Sehr geehtrer Herr Johner,
vielen Dank für Ihren Artikel!
Ein Akteur hat nach Artikel 16 Absatz 1 Herstellerpflichten bei „Bereitstellung eines Produkts auf dem Markt unter dem eigenen Namen, dem eigenen eingetragenen Handelsnamen oder der eigenen eingetragenen Handelsmarke“.
Würde das nicht bei jedem Produzenten einer Behandlungseinheit zutreffen, da dieser verpflichtet ist den eigenen Namen, dem eigenen eingetragenen Handelsnamen oder der eigenen eingetragenen Handelsmarke anzubringen (Annahme: Es liegt keine Vereinbahrung vor)?
Freundlichste Grüße,
Daniel Gostner
Sehr geehrter Herr Gostner,
das ist eine super Frage!
Der Inverkehrbringer der Behandlung ändert üblicherweise nichts an den einzelnen Produkten der Behandlungseinheit. Vielmehr erstellt er eine zusätzliche Verpackung, die die unveränderten Produkte zu eben dieser Behandlungseinheit zusammenfügt.
Beste Grüße, Christian Johner
Hallo,
kurze Anmerkung zum o. g. Abschnitt 2.f.), resultierend aus einem vermeintlichen Übersetzungsfehler:
Artikel 16(4)
„Der Händler – und nicht der Hersteller – (oder Importeur) muss der zuständigen Behörde eine Bescheinigung vorlegen, (…) dass das Qualitätsmanagementsystem des Händlers oder Importeurs den in Absatz 3 festgelegten Anforderungen entspricht.“
Lt. dem englischen Originaltext (Artikel 16(4), 2. Satz) ist lediglich der Händler (und Importeur) angesprochen, mit der zuständigen Behörde zu kommunizieren.
Viele Grüße
M. Herzog
Danke für den wertvollen Hinweis, Herr Herzog!
Sie haben Recht: Auf die deutsche Variante der MDR kann man sich leider nicht verlassen.
Der Artikel ist bereits geändert.
Viele Grüße, Christian Johner
Guten Tag Herr Prof. Johner,
vielen Dank für Ihren zusammenfassenden Artikel!
Ein Händler muss nur über ein Qualitätsmanagementsystem verfügen, wenn er Tätigkeiten entsprechend §16 ausübt. Ansonsten muss er kein QMS unterhalten. Ihrem Fazit ist zu entnehmen, dass der Händler prinzipiell über ein QMS verfügen muss?!?
Viele Grüße
Martina Feldmann
Sehr geehrte Frau Dr. Feldmann,
die Schlussfolgerung, dass ein Händler ein QMS unterhalten sollte (nicht muss!), auch wenn der Händler keine der in §16 genannten Tätigkeiten ausübt, zieht die irische Behörde. Rechtlich einfordern lässt sich dies in diesen Fällen aber nicht.
Allerdings sollten die Hersteller Wert darauf legen, dass die Händler beispielsweise die Rückmeldungen einem Prozess unterwerfen.
Beste Grüße, Christian Johner
Hallo Herr Prof. Dr. Johner,
es wird von Ihnen ausgeführt, dass Händler „… Produkte sogar ‚umkennzeichnen‘ und ‚umverpacken‘ …“ dürfen, ohne Erlaubnis durch den Hersteller. Dieser muss lediglich darüber unterrichtet werden. Nach meinem Kenntnisstand gilt das nicht für das Relabeln der Artikel. Originallabel der Hersteller dürfen nicht einfach entfernt oder überklebt werden. Wird dies beabsichtigt, benötigt der Händler eine entsprechende Vereinbarung mit dem Hersteller. Oder gilt dies nicht mehr? In unseren Audits wurde und wird von den Auditoren immer sehr viel wert auf diese Vereinbarungen und deren Inhalt gelegt.
Herzliche Grüße
Markus Wand
Sehr geehrter Herr Wand,
die Begriffe des „umkennzeichnens“ und des „umverpackens“ stammt aus der MDR selbst (siehe §16(4)). Die Original-Label darf der Händler nicht einfach überkleben, weil das die Konformität des Produkt gefährden würde. Das gilt weiterhin.
Beste Grüße, Christian Johner
Guten Tag Herr Prof. Dr. Johner
Eine überaus spannende Frage hatten wir nun im Audit. Wie geht ein Händler vor mit Reklamationen. Verschiedene Hersteller haben ja Ihre eigene Page um dies zu melden, was aber wenn nicht?
Dies ist als Händler nicht so einfach zu handhaben und da wäre ein Tipp von den Experten hilfreich.
Eine der Fragen ist, wie grenze ich mich ab mit dem Begriff „Reklamation“
Danke für die Unterstützung und freundliche Grüsse
Marcel Bühler
Sehr geehrter Herr Bühler,
die MDR gibt keine Vorgaben dafür, in welcher Form bzw. über welchen Kanal die Händler an die Hersteller melden müssen. Sie verlangt es aber. Sie verpflichtet beide Seiten, diesen Informationskanal zu gewährleisten. D.h. ein Händler müsste vor dem Verkauf des ersten Produkts eine Vereinbarung mit dem Hersteller getroffen haben, wie dieser Kanal beschaffen sein soll (z.B. Medium, Format, Frist, Form, Ansprechpartner etc.).
Die MDR definiert nur den Begriff „Vorkommnis“, nicht aber den Begriff „Beschwerde“. Ich würde mich daher an die ISO 13485 halten:
Beste Grüße, Christian Johner
Hallo Herr Prof. Dr. Johner,
herzlichen Dank!
Grüße zurück
Markus Wand
Guten Tag Herr Johner,
hinsichtlich der Regelung des §16(2) bin ich etwas irritiert. Der Absatz sagt in beiden Fällen, dass das Umverpacken bzw. das Ändern der Kennzeichnung nur erlaubt ist, „falls das für die Vermarktung der Produkte in dem Mitgliedstaat notwendig ist“. In meinem Verständnis, dachte ich geht es da, um nationale Regelungen die möglicherweise erfüllt werden müssen oder Übersetzungen oder ähnliches. Nicht um ein generelle Erlaubnis des „Umverpackens“.
Vielen Dank für eine Antwort.
Mit freundlichen Grüßen
Moritz Tillmann
Sehr geehrter Herr Tillmann,
danke für die Frage! Ich denke, die Antwort hängt davon ab, wie man die „Klammer“ setzt, auf die sich der Nebensatz „die für die Vermarktung des Produkts in dem jeweiligen Mitgliedstaat erforderlich sind“, bezieht. Meines Erachtens nur auf „weiterer Informationen“. Damit ist die Kennzeichnung nicht nur für die von den „Mitgliedsstaaten notwendigen“ erlaubt.
Es gilt auch zwischen dem „Umkennzeichnen“ und dem „Umverpacken“ zu unterscheiden. Auch das „Umverpacken“ ist nicht darauf beschränkt, dass ein Mitgliedstaat dieses Umverpacken erforderlich macht.
Man will generell den Händlern etwas mehr Möglichkeiten geben, weil das OEM-PLM-Konstrukt weggefallen ist (das es eigentlich offiziell noch nie gab).
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
Sie meinten mehrfach, dass eine Zertifizierung von Händlern nicht eingefordert wird. Der englische Text der MDR besagt:
„Within the same period of 28 days, the distributor or importer shall submit to the competent authority a certificate, issued by a notified body designated for the type of devices that are subject to activities mentioned in points (a) and (b) of paragraph 2, attesting that the quality management system of the distributer or importer complies with the requirements laid down in paragraph 3.“
Somit muss bereits der Händler ein Zertifikat von einer benannten Stelle einreichen, die eine entsprechende Akkreditierung hat, welches bestätigt, dass das QMS des Händlers die Anforderungen aus Artikel 16, Paragraf 3 der MDR erfüllt.
Wie soll aus Ihrer Sicht ein Händler an ein solches Zertifikat ohne entsprechende Vorab-Prüfung durch eine benannte Stelle kommen? Ich verstehe zudem die Iren nicht so, dass keine Zertifizierung notwendig ist, sondern dass kein spezieller QMS-Standart vorgeschrieben wird (“ it is not a requirement that a quality system is officially accredited to any specific standard“). Eine 9001 Zertifizierung sollte eigentlich ausreichen für die in der MDR angesprochenen Punkte.
Vielen Dank für Ihre Zeit und Mühe.
Mit freundlichen Grüßen,
Christian Baudis
Sehr geehrter Herr Baudis,
danke für Ihre wichtige Frage! Wir bewegen uns in der Tat in einem etwas noch nicht abschließend geklärtem Terrain.
Noch eine Vorbemerkung für die anderen Leser vorweg: Diese Diskussion bezieht sich nur auf Händler, die bestimmte in Artikel 16 Absatz 2 genannte Tätigkeiten durchführen.
Nun zu Ihrer Frage: Wenn kein offizieller akkredierter Standard verlangt wird (Sie zitieren exakt die relevante Stelle der irischen Behörde), dann sind die Aussagekraft und Bedeutung eines Zertifikats beschränkt. Denn dann gibt es weder eine Prüfnorm noch eine Akkreditierungsstelle, die die Prüforganisation überwacht. Ihre Einschätzung, dass eine ISO 9001 die Anforderung an ein QM-System erfüllt, teile ich. Es ist allerdings wichtig zu betonen, dass dieses QMS die relevanten Prozesse umfassen muss.
Danke auch für Ihre erste Frage: Sie haben Recht: Ich hatte bei der Korrektur des Artikels aufgrund der falschen Übersetzung der MDR einen Satz übersehen gehabt. Dieser ist nun korrigiert und durch einen Hinweis auf die Unklarheit ergänzt.
Herzlichen Dank dafür!
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
beim Umpacken in Artikel 16 wird von der „äußeren Verpackung“ gesprochen. Die genaue Definition dafür bleibt die MDR meines Erachtens nach schuldig.
Wenn ein Händler ein Set eines Verbrauchsmaterials „anbricht“, indem jeder einzelne Artikel für sich vollständig konform gelabelt und verpackt ist, ist das dann ein Ändern der äußeren Verpackung nach Artikel 16? Selbst wenn der Artikel genau in der Ausführung mit identischem Label neben dem Set auch einzeln auf dem Markt bereitgestellt wird?
Viele Grüße und einen schönen Abend
Dustin Bargfrede
Sie haben Recht, sehr geehrter Herr Bargfrede,
die Definition fehlt. Bei der Antwort muss man sich wahrscheinlich vom Sinn des Gesetzes her nähern: Man möchte verhindern, dass durch die „Umverpackung“ d.h. Umgestaltung der Verpackung Risiken entstehen oder die Überwachbarkeit im Markt leidet. Ein Zerlegen eines Sets in Teile, die bereits in genau dieser Form legal in den Verkehr gebracht werden, dürften keine dieser Risiken verursachen. Allerdings müsste das Thema UDI beleuchtet und analysiert werden, welche UDI auf den einzelnen Verpackungsebenen angebracht ist.
Die Transportverpackungen sind von den Überlegungen ausgenommen.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
wir lassen ein Produkt in Taiwan unter unserem eigenen Handelsnamen herstellen. Auf der Verpackung sind wir als (gesetzlicher) Hersteller angegeben. Gemäß MDR sind wir dann zusätzlich Importeur, ist das richtig? D.h. wir müssen uns auf der Verpackung zusätzlich als Importeur angeben? Wenn ja, auf welcher Verpackungsebene ist diese Angabe notwendig? Reicht die Außenverpackung, oder ist die Angabe des Importeurs auch auf der Unterverpackung notwendig?
Müssen wir in diesem Fall auch überprüfen, ob das CE-Kennzeichen angebracht wurde, eine Konformitätserklärung ausgestellt wurde, etc. Das macht doch keinen Sinn, wenn wir ohnehin als Hersteller angeführt sind und die Verantwortung für das Produkt übernehmen, oder?
Vielen Dank!
Beste Grüße,
Karl-Heinz
Sehr geehrter Herr Suddeara,
die Antwort, nach der Sie wahrscheinlich suchen, findet sich in Artikel 13 Absatz 3. Dort heißt es:
Wenn Sie der Importeur sind (d.h. keinen anderen Importeur bestimmt haben), müssen sie die Angaben auf dem Produkt oder dessen Verpackung anbringen. Sie können sich nicht darauf beschränken, nur die oberen Verpackungsebenen damit zu versehen.
Das Sinn des Gesetzes besteht darin, dass man den Importeur kontaktieren kann, wenn man beispielsweise als Anwender auf Probleme mit dem Produkt stößt. Da man als Anwender aber oft nur das Produkt oder dessen direkte Verpackung zu Gesicht bekommt, wäre eine Angabe auf anderen Verpackungsebenen wenig zielführend.
Sie müssen die CE-Kennzeichnung prüfen. Wenn Sie gleichzeitig Hersteller und Importeur sind, bedeutet das aber nicht, dass Sie das redundant machen müssen. Sie verfügen ja über Aufzeichnungen, die nachvollziehen lassen, dass Sie die korrekte Kennzeichnung geprüft haben.
Die Verantwortung liegt streng genommen (auch) beim Bevollmächtigten. Sie haben wahrscheinlich auch diese dritte Rolle inne.
Beste Grüße, Christian Johner
Vielen Dank Herr Johner für die rasche Antwort.
Ich bin mir leider immer noch nicht ganz sicher, ob wir tatsächlich Importeur des Produkts sind. Das Produkt wird in Taiwan ausschließlich für uns (mit unserer Handelsmarke) von einer Drittfirma hergestellt (produziert). Am Etikett geben wir diese Firma als Produktionsstätte an. Unsere Firma (Sitz in der EU) ist neben dem Herstellersymbol angegeben. Wir führen die Konformitätsbewertung durch und die technische Dokumentation ist ebenfalls bei uns. Ich sehe es so, dass wir Hersteller (in der EU) sind und somit weder ein Bevollmächtigter noch ein Importeur benötigt wird. Ist das richtig?
Sorry, aber irgendwie blicke ich da noch nicht richtig durch…
Beste Grüße,
Karl-Heinz
Sehr geehrter Herr Suddera,
so wie Sie es schildern, sind Sie in der Tat nicht der Importeur, weil Sie selbst als Hersteller fungieren. Möglicherweise ist das eine PLM-OEM-Konstellation, wie Sie bald aussterben wird. Möglicherweise findet die Produktion unter Ihrem QM-Dach statt. In beiden Fällen wäre Sie aber der europäische Hersteller, der für die Konformität gerade steht. Die Importeur im Sinne der MDR bliebe Ihnen auch erspart.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
vielen Dank für Ihre Hilfe und Ihre Mühe!
Beste Grüße,
Karl-Heinz
Sehr geehrter Herr Johner,
in unserem Bereich ist es nicht unüblich das Händler/Homecarer für die Versorgung ihrer Kunden sterile Medizinprodukte aus der Sekundärverpackung auseinzeln und neu sekundär umverpackt an den Kunden abgeben. Wie bewerten Sie diese Tätigkeit unter den neuen Vorgaben der MDR für Händler?
Ist das Anbrechen der Sekundärverpackung bereits nach Artikel 16 (2) b) eine beeinträchtigung des Originalzustandes?
Schöne Grüße
Gerald
Sehr geehrter Herr Gerald,
ich vermute, dass es sich bei der Sekundärverpackung nicht um die Transportverpackung handelt. Wenn dem so wäre, dann ist ein „Auseinzeln“ eine Tätigkeit, die vom Artikel 16 adressiert wird und Ihnen Herstellerpflichten auferlegen würde.
Das ist ggf. nachvollziehbar, wenn man in Betracht zieht, dass der Hersteller mit der Sekundärverpackung Risiken beim Transport, ggf. Probleme mit der Sterilität und der Haltbarkeit adressieren und auf der Verpackung Informationen zum korrekten Umgang geben will. Auch die Vergabe von UDIs muss hier klar geregelt sein.
Wenn Sie zum Ergebnis kommen, dass keine Risiken bestehen, wäre das das Ergebnis einer Risikoanalyse (und ggf. nachzuweisen), nicht aber die Apriori-Annahme.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Dr. Johner,
nachdem nun bei uns das erste OEM-PLM-Verhältnis in ein Hersteller-Händler-Verhältnis übergehen soll, kam bei der Vertragsbesprechung folgende Fragestellung auf: Welche vertraglichen Regelungen und vor allem zwischen welchen Parteien sind notwendig, wenn das Produkt durch den Händler (ehemaliger PLM), unter dem Handelsnamen unseres Händlers auch über weitere Händler in der EU verkauft werden soll? Müssen wir als Hersteller mit jedem Händler unseres Händlers eine vertragliche Regelung eingehen? Wenn dies wirklich so wäre, wäre dies ja ein unheimlicher zusätzlicher bürokratischer Aufwand, da bei mehreren ehemaligen PLMs meistens noch weitere Händler dazu kommen würden. Und dann könnte es ja theoretisch sein, dass ein Händler unsere Produkte unter zwei Handelsnamen (zwei „Zwischenhändler“) verkauft.
Vielen Dank für Ihren Support!
Herzliche Grüße
K. Lippert
Sehr geehrter Herr Lippert,
danke für diese wirklich wichtige Frage!
Die kurze Antwort auf Ihre Frage ist: Ich würde Ihnen in der Tat empfehlen, einen Vertrag mit Ihren Händlern abzuschließen. Darin könnten Sie beispielsweise regeln:
Ich sehe für Sie eher eine Chance als nur den Aufwand.
Herzliche Grüße, Christian Johner
Sehr geehrter Herr Johner,
wir sind noch ganz neu auf dem Gebiet –
Gilt oben stehender Artikel auch komplett bei der neuen Richtlinie die jetzt erst ab Mai 2021 in Kraft treten wird?
Vielen Dank
C. Apciella
Sehr geehrte Frau Apiciella,
Danke für Ihre Frage! Ich bin noch nicht ganz sicher, ob ich sie verstehe. Die Richtlinie (MDD) läuft aus, die Verordnung (MDR) tritt im Mai 2021 in Kraft. Von Übergangsfristen abgesehen, gilt die MDR dann in ganzem Umfang. Für Händler gibt es keine Übergangsfristen.
Wenn meine Antwort nicht zu Ihrer Frage passt, dann haken Sie einfach nach.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
sie schreiben:
„Wenn jedoch ein Händler die Produkte von einem Hersteller oder einem anderen Händler einkauft, der nicht in der EU angesiedelt ist, übernimmt dieser Händler zusätzlich die Rolle des Importeurs. Die MDR definiert Importeure wie folgt:
„„Importeur“ bezeichnet jede in der Union niedergelassene natürliche oder juristische Person, die ein Produkt aus einem Drittland auf dem Unionsmarkt in Verkehr bringt;“ Quelle: MDR, Artikel 2“
In diesem Zusammenhang sollte aber noch die Definition des „Inverkehrbringens“ unter die Lupe genommen werden. Die MDR definiert das Inverkehrbringen wie folgt:
„„Inverkehrbringen“ bezeichnet die erstmalige Bereitstellung eines Produkts, mit Ausnahme von Prüfprodukten, auf dem Unionsmarkt“ Quelle: MDR, Artikel 2
Demnach wird zum Importeur nur derjenige Händler, der ein Produkt aus einem Drittland auf dem Unionsmarkt erstmalig in Verkehr bringt. Wurden diese Produkte von einer anderen Partei bereits auf dem Unionsmarkt bereitgestellt (vom anderen Händler oder Bevollmächtigten), so können sie vom Händler bezogen und vertrieben werden, ohne dass er zum Importeur wird, oder?
Liebe Grüße
Jacek Cecek
Genauso ist es, Herr Cecek!
Sehr geehrter Herr Prof. Johner,
vielen Dank für die sehr hilfreiche Übersicht.
Ein Sachverhalt erschließt sich mir leider noch nicht.
Wie unterscheiden sich die Pflichten eines Händlers und Zwischenhändlers?
Nehmen wir folgende Lieferkette:
Der Hersteller lizensiert ein Medizinprodukt an den Partner X. Dieser bewirbt das Medizinprodukt und nennt seinen Namen zusätzlich zum Hersteller auf der Packung. Das Medizinprodukt wird A. über den Pharmazeutischen Großhandel in die Apotheke verkauft. Die Apotheke hält es vorrätig und gibt zur Inbetriebnahme an Patienten bei Bedarf ab. Das Medizinprodukt wird B. direkt an Krankenhäuser geliefert, in denen das Medizinprodukt in Betrieb genommen wird.
Ist Fall A wäre Partner X ein Zwischenhändler und bei B ein Händler? Andern sich die Verpflichtungen?
Ich freu mich auf Ihre Antwort! Vielen Dank!
Sehr geehrte/r M.W.,
die MDR unterscheidet Händler und Zwischenhändler nicht. Ein Händler ist definitionsgemäß
Sowohl der Händler als auch der Zwischenhändler entsprechen dieser Definition. Damit haben beide die gleichen Verpflichtungen. Das ist auch nicht ungewollt. Man will über die komplette Lieferkette das Produkt nachverfolgen können und sicherstellen, dass alle Beteiligten z.B. an der Kommunikation von Zwischenfällen aktiv mitwirken.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
trifft „gefälschtes Produkt“ auch zu, wenn ein Hersteller ein MP als MP einstuft, obwohl dieses KEIN MP ist oder gibt es hierfür eine andere Bezeichnung?
Vielen Dank
Daniel
Sehr geehrter Daniel, sehr geehrter Herr,
danke für Ihre kurze und präzise Frage.
Eine Fälschung würde man das nicht nennen. Das ist eine Fehlqualifizierung. Qualifizierung ist die Entscheidung, ob ein Produkt ein MP ist. Auch eine Fehlqualifizierung ist gesetzeswidrig.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
vielen Dank für Ihre schnelle Antwort bezüglich „gefälschtes Produkt“.
Gibt es in der MDR oder in einer sonstigen VO/RL etc. explizit einen Passus, auf welchen man sich beziehen kann bezüglich falscher Einstufung/Qualifizierung? Ich konnte leider nichts finden.
Derzeit sind Amalgamabscheider (trennen Amalgam vom Abwasser) als MP (Klasse I) eingestuft. Ich und einige Kollegen von mir sehen das nicht so. Das Argument eines Herstellers ist, dass ein Abscheider Krankheiten verhütet (präventiv), womit der Hersteller nicht ganz unrecht hat aber dann müsste ein Fahrzeugkatalysator ebenfalls ein MP sein oder nicht?
Vielen Dank für Ihre Hilfe und auch für Ihre unglaublich Hilfreiche Website.
Daniel
Sehr geehrter Daniel,
abhängig von der Richtlinie bzw. Verordnung greifen das MPG bzw. das MPDG. Dort gibt es Strafvorschriften.
Es kommen aber auch weitere Gesetze in Betracht wie das Wettbewerbsrecht.
Im konkreten Fall würde ich den Amalgamabscheider auch nicht als Medizinprodukt qualifizieren. Meines Erachtens steht hier als Zweck nicht der Verhütung von Krankheiten im Vordergrund, sondern die Erfüllung von Umweltauflagen. Jedoch wäre eine Qualifizierung als Zubehör zu Dentalprodukten durchaus denkbar.
Ein Fahrzeugkatalysator wird üblicherweise als Teil eines Autos in Verkehr gebracht. Alleine kann er keine Krankheiten verhüten.
Eine Klage gegen die Qualifizierung hätte eine nicht verschwindend geringe Wahrscheinlichkeit eines Erfolg.
Viele Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
im Artikel 14 werden die allgemeinen Pflichten der Händler beschrieben.
Ist es, nach der ersten Kontrolle von bezogenen Produkten vom Hersteller und des erfolgreichen Nachweises der Einhaltung der Anforderungen seitens des Händlers notwendig, tatsächlich eine 100%ige WE-Kontrolle durchzuführen?
Normalerweise sollte die Identprüfung, Mengenprüfung, Labelingcheck, LOT-Nummer und Verfallsdatum ausreichen.
Dies kann natürlich zu 100% umgesetzt werden – wenn aber eine Gebrauchsanweisung der Verkaufsverpackung beiliegt, kann ich das so nicht prüfen – gleichermaßen ein Label auf dem Einzelprodukt.
Würde dann der Check pro Verpackungseinheit, so wie sie zum Kunden geht, als 100% WE-Kontrolle ausreichen?
Bei einem Probenahmeverfahren müsste die Verpackung geöffnet werden – könnte sogar bis in den Bereich der Aufhebung der Sterilität geben, wenn ich zusätzlich zum Labeling auf der Verpackung das Label am Produkt prüfen muss.
U.u. kann ich Produkte nach der Prüfung nicht mehr in Verkehr bringen – das kann nicht im Sinne der Verordnung sein.
Ist es möglich, durch eine Qualitätssicherungsvereinbarung mit dem Hersteller wesentliche Teile der WE-Prüfung durch den Hersteller bei der WA-Prüfung durchführen zu lassen.
Sehe Ihrer Antwort mit Interesse entgegen.
Karl-Heinz Sommerlade
Sehr geehrter Herr Sommerlade,
danke für Ihre wichtige Frage!
Es gibt keine Verpflichtung der Händler zu einer 100% Wareneingangskontrolle. Das gilt auch nicht für das Labeling. Die MDR schreibt explizit:
Die CE-Kennzeichnung und die UDI müssten sie von außen erkennen können. Dass eine Gebrauchsanweisung innerhalb eines sterilisierten Produkts verpackt wird, ist mir nicht bekannt. Daher sehe ich da kein Problem.
Es kann allerdings vorkommen, dass Sie stichprobenartig Produktverpackungen öffnen müssen, um zu prüfen, ob die Gebrauchsanweisung beiliegt. Das wäre aber Stichproben. Hier könnten Sie den Hersteller bitten, entsprechend geöffnete Produkte zurückzunehmen.
Für wichtig würde ich es halten, dass Sie im Rahmen der Post-Market Surveillance systematisch prüfen, ob von Kundenseite Probleme gemeldet werden. Da würde das Fehlen einer Gebrauchsanweisung dazuzählen.
Mit den besten Grüßen, Christian Johner
Sehr geehrter Herr Professor Johner,
mir ist immer noch nicht ganz klar, ob man als Händler über ein QMS verfügen muss. In der deutschen Übersetzung heißt es:
„Der Hersteller oder Importeur legt der zuständigen Behörde im selben Zeitraum von 28 Tagen eine Bescheinigung vor, ausgestellt von einer Benannten Stelle und bestimmt für die Art der Produkte.“
Sie sagen, „Hersteller“ wäre hier ein Übersetzungsfehler und sollte eigentlich „Händler“ heißen. Nun sagt aber die MDR in „ANHANG XII; VON EINER BENANNTEN STELLE AUSGESTELLTE BESCHEINIGUNGEN“ unter Punkt 3:
Die Bescheinigungen werden nur an einen Hersteller ausgestellt.
Warum sollte ein Händler ein QMS-System von einer benannten Stelle zertifizieren lassen?. Selbst wenn er ein QMS hat, würde nicht auch ein Zertifizierer ausreichen, muss es eine benannte Stelle sein?
Und gleich ein Frage im Anschluss:
Muss die Vorgehensweise in Artikel 16 (4) bei jeder Produktcharge die verkauft wird eingehalten werden oder nur beim erstmaligen Verkauf dieses Produkts?
Vielen Dank für Ihre Hilfe
Walter Breuer
Sehr geehrter Herr Breuer,
bei dem Artikel, den Sie zitieren, geht es nicht um „normale“ Händler, sondern Händler, die z.B. die Zweckbestimmung des Produkts ändern oder es unter dem eigenen Namen in den Verkehr bringen. Damit übernehmen Sie wie der Artikel besagt Herstellerpflichten. D.h. sie werden wie Hersteller behandelt und daher zumindest für den relevanten Teil auch auditiert. Die MDR spricht explizit von „notified body“ und nicht von Zertifizierer.
Wenn der Händler, der nun auch Herstellerpflichten übernommen hat, ein QMS hat, dann stellt er damit sicher, dass die in dem Artikel weiter o.g. Tätigkeiten konform verlaufen. Damit muss er nicht für jede Charge neu informieren.
Beste Grüße, Christian Johner
Vielen Dank für die schnelle Antwort Herr Professor Johner,
eine Frage hätte ich noch zur Zertifizierung. Nachdem nur die für den Händler relevanten Bereiche auditiert würden, würde aus ihrer Sicht eine Zertifizierung nach ISO 9001 ausreichen oder besser nach ISO 13485?
Beste Grüße
Walter Breuer
Sehr geehrter Herr Breuer,
ich vermute, Ihre Frage bezieht sich wieder auf den gleichen Artikeln der MDR. Falls Sie die Forderung nach einer Benannten Stellen erfüllen müssen, dann sind damit Benannten Stelle gemeint, die QMS zertifizieren dürfen, und das sind die nach ISO 13485.
Beste Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
ich habe soeben folgende Antwort einer renommierten „benannten Stelle“ in Deutschland erhalten:
„Das in Artikel 16 Absatz 4, 2017/745 MDR genannte „Zertifikat“ für Händler oder Importeure ist nicht durch ein ISO13485 Zertifikat abgedeckt.
Der Prüfungsprozess und der Inhalt dieses Zertifikats werden noch auf europäischer Ebene erörtert.
Deshalb bedauere ich, Ihnen mitteilen zu müssen, dass wir noch keine Bescheinigungen gemäß Artikel 16 Absatz 4, 2017/745 MDR ausstellen können.“
Da fragt man sich, wie man die Anforderungen der MDR als Händler erfüllen soll?
Sehr geehrter Herr Professor Johner,
Sie schrieben in einem Kommentar, dass die MDR keine Unterscheidung zwischen Händler und Zwischenhändler macht und dass ein Hersteller im besten Fall mit all seinen Händlern einen Vertrag machen sollte.
Meine Frage:
Ein Händler (und meiner Meinung nach ist er auch der Importeur) möchte uns ein Produkt zum Weitervertrieb verkaufen und sieht selbst aber keinen Grund eines Vertrages zwischen uns. Mit dem Hersteller selbst stehen wir nicht in Kontakt. Der Händler steht auf dem Label (jedoch nicht explizit als Importeur) – wir natürlich gar nicht. Sollten wir aktiv auf den Hersteller zwecks Vertrag zugehen? Welche Pflichten hätten wir, wenn wir nun eine zusätzliche Vertriebsetikette und ggf. Umverpacken würden? Wir stehen ja wie gesagt gar nicht im Kontakt mit dem Hersteller.
Ich hoffe, Sie können mir helfen. Vielen Dank dafür im Voraus.
Beste Grüße
Antonia Kamp
Sehr geehrte Frau Kamp,
wie Sie erwähnen unterscheidet die MDR nicht zwischen Händler und Zwischenhändler und Endhändler. Wenn Sie in die Klasse fallen, müssen Sie die Anforderungen u.a. des Artikels 14 erfüllen.
Dieser verpflichtet Sie, die Hersteller und „gegebenenfalls den Bevollmächtigten des Herstellers und den Importeur“ bei Problemen zu informieren. Einen Vertrag benötigen Sie deshalb mit diesen Organisationen nicht. Eigentlich sollten diese Sie darum bitten.
Stellen Sie aber sicher, dass die Kommunikationswege zu diesen Organisationen funktionieren, damit Sie Ihrer Pflicht nachkommen können.
Beste Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
wir kaufen Komponenten von Herstellern ein und verkaufen diese dann als Händler an unsere Kunden weiter.
Da es durch die MDR kein OEM-PLM-Konstrukt mehr gibt, stehen wir aktuelle vor der Frage der Kennzeichnung von Produkten, die durch mehrere Händler vertrieben werden.
Wie müssen wir Produkte kennzeichnen bei denen wir als Zwischen-Händler für unsere „OEM“-Kunden auftreten?
Müssen durch die MDR nun beide Unternehmen als Händler auf dem Produkt stehen oder nur der End-Händler – also unser „OEM“-Kunde, der „seine“ Produkte nun als Handelsware vertreibt?
Ich hoffe Sie können uns hier helfen.
Vielen Dank im Voraus.
Mit freundlichen Grüßen,
A. Raabgrund
Sehr geehrte Frau Raabgrund,
das sind spannende Fragen!
Sie schreiben, dass Sie Komponenten kaufen. Ich vermute daher, dass dies keine Medizinprodukte sind.
Von den PLM-OEM-Konstrukten sprach man „nur“ im Kontext der Medizinprodukte (einschließlich Zubehör). Es ging dabei um die Frage, welche Anforderungen an die Inverkehrbringer gelten, die nicht der tatsächliche Hersteller der Medizinprodukten sind.
Bei Komponenten (Ersatzteilen?) sieht das anders aus. Hier greifen die etwas niederigen Anforderungen des Artikels 23 der MDR.
Die MDR definiert den Begriff des Händlers wie folgt:
Produkte wiederum definiert die MDR als „Medizinprodukte und ihr Zubehör sowie die in Anhang XVI aufgeführten Produkte …“.
Selbst wenn Sie (nur) mit den Komponenten handeln, sind Sie im Sinne der Verordnung kein Händler. Insofern entfallen die entsprechenden Pflichten — zumindest bezüglich der „Produkte“.
Beste Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
soweit ich es verstanden haben gelten die Händler- und Importeurspflichten unabhängig von den Abverkaufsfristen bereits ab dem Geltungsbeginn der MDR am 26.05.21. Heißt dieses, dass wir z. B. in der Rolle als Importeur Ware, die wir nach diesem Termin erhalten, unabhängig davon, ob es sich um Legacy Produkte oder bereits nach MDR zertifizierte Produkte handelt, mit den Informationen zum Importeur kennzeichnen müssen oder bezieht sich dieses nur auf bereits MDR zertifizierte Produkte? Was ist mit bestehendem Lagerbestand, muss dieser auch gekennzeichnet werden?
Grüße
H. Simon
Sehr geehrter Herr Simon,
das ist eine spannende Frage. Nach unserem Verständnis hängen die Pflichten der Wirtschaftsakteure (Importeure/Bevollmächtigter/Händler) am Produkt. D.h., wenn ein Produkt noch nach MDD legal in Verkehr gebracht wird, muss derjenige erstmal „nur“ die MDD-Forderungen erfüllen, ausgenommen der Registrierungsanforderungen gemäß MDR/EUDAMED. Dies wurde uns von anwaltlicher Seite und auch durch ein Regierungspräsidium bestätigt. Eine Guidance oder ein Gerichtsurteil wären für diesen Fall dennoch sicherlich hilfreich, um eine definitive Aussage treffen zu können.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Herr Professor Johner,
auf die Frage von Herrn Sommerlade nach Artikel 14 / WE-Kontrolle führten Sie (wie in der MDR ausgeführt) an, dass für die nach UnterAbsatz 1 genannten Buchstaben a, b und d keine 100% Wareneingangskontrolle für die Händler erforderlich ist bzw. hier das Stichprobenverfahren angewendet werden kann.
Bedeutet das im Umkehrschluss, dass ab dem Importeur jeder Groß.- / Einzelhändler bzw. Apotheker bei Importprodukten (Buchstabe c) eine 100 % Kontrolle (Packung für Packung) des Wareneingangs durchführen muss?
Müssten dann z.B. Apotheken die Diabetes-Zentren beliefern, jede Packung Teststreifen oder Nadeln einzeln prüfen?
Vielen Dank und viele Grüße
Sehr geehrter Herr Thiel,
danke für Ihre Frage!
Sie verweisen auf die Pflicht der Händler zu überprüfen, ob die Importeure ihren in Artikel 13 (3) genannten Pflichten gerecht werden. Das ist die Pflicht, die Angaben zum Importeuer auf der Verpackung bzw. den Begleitmaterialien anzugeben.
Weshalb die MDR hier das Stichprobenverfahren nicht erlaubt, entzieht sich unserer Kenntnis. Ich vermute, dass es damit zusammenhängt, dass der Importeur bei seiner Überprüfung kein Stichprobenverfahren anwenden darf (s. Art 13). Deshalb haben die Autoren vermutlich Punkt c) ausgenommen. Vermutlich soll diese erweiterte Prüfung durchgeführt werden, um Fälle bei denen Importeure nicht sorgfältig arbeiten und z.B. nicht alle Produkte zusätzlich kennzeichnen bzw. die notwendigen Informationen beilegen, zu reduzieren.
Herzliche Grüße, Luca Salvatore
Sehr geehrter Herr Johner,
unsere Firma möchte die berühmten Coronaselbstests von einem EU-Rep beziehen und handeln.
Zum Thema Meldepflicht bin ich mir, nach Recherche im MPSV (Artikel 3, Absatz 3) nicht ganz sicher, ob wir dem BfArm UND dem EU-Rep/Importeur Vorfälle melden müssen.
Unter 2d) sprechen Sie von „Beschwerden sowie Berichte über Vorkommnisse sammeln und an die Hersteller und ggf. Importeure weiterleiten.“
Wird ein Händler (aktuell und unter MDR) also generell nur an den Importeur melden müssen bei Vorkomnissen?
Beste Grüße
BD
Sehr geehrter Herr D.
das MPSV ist nur noch kurze Zeit gültig. Danach „übernimmt“ die MDR und die MPAMIV. Als Händler müssen Sie gemäß Artikel 14 Absatz (2) abhängig vom Schweregrad dem Importeuer, dem Händler, dem Bevollmächtigten und der Behörde melden.
Bis Mai sind das MPG und die MPSV gültig. Solange muss der Händler an den Hersteller bzw. EU-Rep melden. Allerdings kann es sein, dass der Importeuer in die Verantwortung genommen wird, wenn er ohne EU-Rep importiert.
Der Beitrag bezieht sich aber auf die MDR, nicht auf die alte Gesetzgebung. Da die MDR kurz bevorsteht, ist das für Sie wahrscheinlich auch die relevantere Information.
Viele Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
wie bewerten Sie folgende Sachlage.
Ein Hersteller (A) stellt außerhalb der EU seine Medizinprodukte her und hat auch dort seinen Hauptsitz.
Eine unselbstständige oder auch selbstständige Niederlassung (B) importiert die Produkte in die EU. Ein Händler (C ) bestellt und erhält die Produkte von (B).
Ist (B) nun seinerseits der 1. Importeur, gemäß Artikel 13 MDR oder ist in dem Fall die Niederlassung ein „verlängerter“ Arm von (A) und der erste Nicht-Hersteller in der Kette ist der Händler (C ) und damit dieser auch gleich 1. Importeur?
Andererseits erfüllt (B) ja auch die Definition als 1. Importeur …
Mit freundlichem Gruß
Andreas Keller
Sehr geehrter Herr Keller,
danke für Ihre Frage!
Wenn die Niederlassung eine in der EU niedergelassene natürliche oder juristische Person ist, welche ein oder mehrere Produkte von A auf dem Unionsmarkt in Verkehr bringt, dann ist die Niederlassung per definitionem ein Importeur. Es gibt keine Unterscheidung, ob diese Niederlassung selbstständig ist oder nicht. Wichtig ist, ob diese Person in der EU niedergelassen ist.
Durch Verträge sollte klar gemacht werden, wer welche Pflichten hat. Die könnten z.B. so gestaltet werden, dass gar nicht die Niederlassung, sondern der Händler die Produkte in den Verkehr bringt. Dann wäre die Niederlassung kein Importeuer. Eine Organisation kann auch mehr als eine Rolle einnehmen.
Bitte beachten Sie auch, dass es noch eine sehr wichtige Rolle gibt, nämlich die des EU-Bevollmächtigten. Hierzu würde sich eher die Niederlassung als der Händler anbieten.
Viele Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
vorab großen Dank für den ausführlichen Artikel. Ich habe folgende Thematik: Wir lassen uns zum einen als Händler und als Hersteller für Software zertifizieren. Später soll noch der Auftakt als Hersteller für ein physisches Medizinprodukt folgen. Ein entsprechendes Qualitätssystem ist als Händler sowie als Hersteller für Medizinsoftware in Erstellung. Ich würde gerne wissen ob es Sinn macht, ein Risikomanagement für alle 3 genannten Rollen zu vereinen oder ob diese getrennt gehalten werden sollten. Als Händler fällt unter anderem schon mal die gesamte Lebenszyklus Evaluierung weg, als Hersteller für Software lehne ich mich zusätzlich zur ISO14971 auch an IEC 62304. Als Hersteller für ein Medizinprodukt dass keine Software ist, nur die ISO14971. Hinzu kommt die Entscheidung für eine geeignete Bewertungsmethode – ich sehe keine Möglichkeit EINE Methode für alle 3 Rollen zu etablieren. Würden Sie demnach empfehlen EIN Risikomanagement für alle 3 Rollen zu etablieren oder diese getrennt zu halten?
Herzliche Grüße,
Nadine Snopek
Sehr geehrte Frau Snopek,
danke für Ihre anspruchsvolle Frage!
Wenn Sie davon sprechen, sich als Händler und als Hersteller zertifizieren zu lassen, dann gehe ich davon aus, dass Sie eine Zertifizierung Ihrer Organisation nach ISO 13485 meinen, wobei das QM-System Ihre Tätigkeiten als Händler und Hersteller im QM-System beschreibt. Falls dem nicht so wäre, kann es sein, dass meine Antwort nicht zielführend ist.
Sobald Sie ein QM-System etablieren, müssen Sie für alle darin beschriebenen Prozesse die Risiken betrachten. Dabei können Sie sich zwar von der ISO 14971 leiten lassen. Die ISO 14971 hat aber als Anwendungsbereich die Hersteller.
Was Sie mit „Methode“ meinen, verstehe ich nicht ganz. Die Methoden des Risikomanagements insbesondere der Risikoanalyse unterscheiden sich aber durchaus: Während beispielsweise bei einem Produkt eine dFMEA Anwendung findet, sind es bei Prozesse eher die pFMEAs. Eine pFMEA eignet sich allerdings für einen Produktionsprozess genauso wie für einen Prozess eines Händlers.
Es gibt nicht „EIN Risikomanagement“. Es gibt Produkte und Prozesse, für welche die Risiken identifiziert und beherrscht werden müssen. Wesentlich ist auch die Entscheidung, ob es ein oder mehrere Zertifikate gibt. Wenn die zertifizierte Organisation im Wesentlichen deckungsgleich wäre, spricht das für eine Zertifizierung.
Falls ich Ihre Situation nicht richtig erfasst haben sollte, dann haken Sie gerne nach.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
Ich habe eine Frage zu Artikel 14 Absatz (2) b:
Muss ein Händler lediglich prüfen, ob die Kennzeichnung und Gebrauchsanweisung in einer oder mehreren festgelegten Sprachen beiliegen oder muss ein Händler grundsätzlich prüfen, ob die Hersteller die Informationen gemäß Anhang I Abschnitt 23 überhaupt bereitgestellt haben? Letzteres würde ja bedeuten, dass man als Händler umfassende Produktkenntnisse haben muss, um dies beurteilen zu können.
Vielen Dank und viele Grüße
Sarah Dreßen
Sehr geehrte Frau Dreßen,
als Händler müssen Sie die Existenz der Materialien prüfen, nicht deren Korrektheit.
Es ist genau wie Sie sagen: Die Überprüfung der Korrektheit setzt Produktkenntnisse voraus.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Dr. Johner,
könnten Sie mir bitte bei der Interpretation des Artikels 16 der MDR behilflich sein.
Im Absatz 2 steht, dass Übersetzungen der bereitzustellenden Informationen nicht als eine Änderung angesehen werden, die Auswirkungen auf die Konformität des Produkts haben.
Im Gegenzug dazu steht in Absatz 4 des gleichen Artikels, dass der Händler bei einer solchen Tätigkeit den Hersteller und die zuständige Behörde vor der Absicht informieren muss UND dass eine Bescheinigung EINER Benannten Stelle der Behörde vorgelegt werden muss, die für die Art der Produkte und die sich über die Tätigkeiten der in Absatz 2 a+b genannten Tätigkeiten erstreckt.
Meine Gedanken dazu:
Auch bei Ihnen im Blog lese ich, dass der Händler ein QMS benötigt, aber dies nicht zwangsläufig zertifiziert sein muss – wie bringe ich das nun mit einer Benannten Stelle in Vereinbarung?
Eine Benannte Stelle kommt bislang für mich immer ins Spiel, um Prüfungen und Bewertungen im Rahmen der vom Hersteller durchzuführenden Konformitätsbewertung durchzuführen. Nach Absatz 2 gibt es aber eigentlich gar kein Problem mit der Konformität. Was soll also jetzt EINE Benannte Stelle da?
Anfangs dachte ich, dass es sich dabei um die Benannte Stelle des Herstellers handelt, die mit dem Hersteller zusammen Überwachungstätigkeiten vornehmen muss, um diesen ausgelagerten Prozess zu überwachen, mittlerweile ist mein Informationsstand der, dass dies wohl nicht so ist und es sich um eine eigens vom Händler beauftragte Benannte Stelle handelt? Ist das richtig?
Wenn ja, was bedeutet das für Händler, die
a) ein QMS haben, aber nicht nach ISO 13485 oder ISO 9001 zertifiziert sind? Haben diese Händler überhaupt eine Chance eine Benannte Stelle für solche „Pseudozertfizierungen“ zu finden? Kann man etwas zu den Kosten sagen (im Vergleich zu einer ISO 13485 Zertifizierung)?
b) ein zertifiziertes QMS nur nach ISO 9001 vorweisen können, also i.d.R. nur eine Zertifizierungsstelle aber keine Benannte Stelle dahintersteht?
c) ein zertifiziertes QMS nach 13485 haben, was bereits durch eine Zertifizierungsstelle zertifiziert wird, welche auch als Benannte Stelle akkreditiert ist? Kann eine Bescheinigung wie in Absatz 4 gefordert, im Zuge der „normalen“ ISO 13485-Zertifizierung ausgestellt werden oder muss eine solche Begutachtung unabhängig davon beauftragt und bezahlt werden?
Vielen Dank
Kirsten Lippert
Sehr geehrte Frau Lippert,
Artikel 16 dreht sich um den Parallelimport. Bei eigenmächtigen Übersetzungstätigkeiten muss der Händler ein QM-System aufbauen, welches mindestens die in Artikel 16 (3) genannten Aspekte umfasst. Es ist in jedem Fall eine separate Auditierung durch eine benannte Stelle notwendig. Dies muss nicht die benannte Stelle des Herstellers sein.
Eine ISO-Zertifizierung ist nicht notwendig, andererseits aber auch nicht hinreichend. Die benannte Stelle wird in jedem Fall ein Audit vor Ausstellung einer Bescheinigung durchführen.
Konkret zu den Fragen:
Aufgrund der Auslastung der benannten Stellen wird es schwierig sein, einen zeitnahen Termin zu bekommen. Die Kosten kenne ich leider nicht.
Reicht wie oben beschrieben nicht aus.
Vermutlich könnte die benannte Stelle ein kombiniertes Audit durchführen und die zusätzlichen Aspekte nach Artikel 16(3) prüfen.
Viele Grüße, Christian Johner und Luca Salvatore, von dem die Antwort stammt.
Dear Prof. Johner und Dr. Luca,
I wanted to ask please if it is possible for the distributor to register in the EUDAMED?
Thank you in advance!
Best regards,
Rania
Dear Rania,
according to ANNEX VI Part A 1.1. EUDAMED requires only
I.e. not all economic operators are registered.
Regards, Christian Johner
Sehr geehrter Prof. Dr. Johner,
vielen Dank für Ihre wertvollen Beiträge.
Wir kämpfen mit der Definition der Rolle des Importeurs / Händlers unter der MDR.
Fallkonstellation: Ein Hersteller aus einem Drittstaat (A) liefert direkt an ein Unternehmen in der EU (B).
Ist B damit zwangsläufig Importeur, da es keinen dazwischengeschalteten Rechtsträger in der EU gibt („Importeur“ iSd MDR)?
Oder könnte die Lieferbeziehung so ausgestaltet werden, dass A das MD in die EU „importiert“ (ohne dass er Importeur iSd MDR wäre) und B z.B. von einem Lager des A innerhalb der EU bezieht, sodass B dann nur „Händler“ iSd MDR wäre? In dieser Konstellation gäbe es dann gar keinen „Importeur“ iSd MDR.
Beste Grüße
Johannes
Sehr geehrter Johannes,
danke für Ihre spannende Frage!
Ein Import ohne die Rolle des Importeurs ist illegal. Daher brauchen wir den Fall nicht weiter zu untersuchen und können uns den Lösungsansätze zuwenden. Die wären beispielsweise:
Viele Grüße, Christian Johner
Sehr geehrte Damen und Herren,
wie sieht die aktuelle Rechtslage (stand heute 30.05.21) bzgl der Kennzeichnungspflicht aus.
Produkt Antigen Schnelltest wird aus China importiert (Deutscher Händler bestellt beim Hersteller in China) Hersteller hat einen EC rep. Produkt hat Sonderzulassung nach §11 MPG.
In der Gebrauchsanweisung, sowie auf der Verpackung stehen die Adressen des Herstellers aus China sowie die Adresse des EC-Rep.
Muss die Adresse des deutschen Händlers ebenfalls mit in die Gebrauchsanweisung UND/ODER (?) auf die Verpackung? Wenn ja, genügt es, dass die Adresse in der Gebrauchsanweisung angegeben ist, oder MUSS diese auch auf die Verpackung?
Vielen Dank.
Sehr geehrter Herr Mueller,
die Anforderungen der MDR finden Sie im Anhang I Abschnitt 23. Die Kennzeichnung muss den Hersteller und den Bevollmächtigten nennen, nicht den Händler. Der Händler muss auch nicht in der Gebrauchsanweisung genannt sein.
Bitte beachten Sie, dass ein Import ohne offiziellen Importeur illegal ist.
Viele Grüße
Christian Johner
Liebes Johner-Team,
Eine Frage zum Anbringen der Adresse des Importeurs.
Wer muss die Information anbringen? Bereits der nicht-EU Hersteller, der nach Europa importiert? Oder genügt es, wenn erst der Importeur in Europa das Etikett anbringt?
Wie steht es bei der Schweiz? EU-Hersteller oder Schweizer Importeur?
Herzlichen Dank und beste Grüße!
Sehr geehrte Frau Mirschberger,
das Labeling ist die Aufgabe und die Verantwortung des Herstellers. Daher darf nicht der Importeur weitere Informationen aufbringen, es sei denn dieser Prozess unterliegt dem QM-System des Herstellers. Das ist auch im Fall der Schweiz so.
Viele Grüße, Christian Johner
Liebes Johner-Team,
Noch eine Frage zum Anbringen der Adresse eines Importeurs:
Ein Lieferant von uns ist der Ansicht, dass es nach MDR Artikel 13 (3) ausreichend ist, den Importeur auf dem Lieferschein anzugeben.
(3) Importeure geben auf dem Produkt oder auf seiner Verpackung oder auf einem dem Produkt beiliegenden Dokument ihren Namen, ihren eingetragenen Handelsnamen oder ihre eingetragene Handelsmarke, ihre eingetragene Niederlassung und die Anschrift an, unter der sie zu erreichen sind, so dass ihr tatsächlicher Standort ermittelt werden kann.
Ist das korrekt? Oder ist nicht eigentlich die IFU mit „einem dem Produkt beiliegenden Dokument“ gemeint?
Herzlichen Dank!
Sehr geehrte Frau Mirschberger,
es ist wie Sie sagen: Das Aufbringen der Händler ist nicht verlangt. Das wäre z.B. bei MPs, die über Apotheken vertrieben werden, auch gar nicht möglich.
Das Dokument kann, muss aber nicht die IfU sein. Es kann auch ein spezielles Dokument für diesen Zweck sein.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
gibt es bezüglich einer (allgemeinen) Registrierung/Registrierungspflicht von Händlern in Deutschland mittlerweile ein Update? Im Artikel sprechen Sie diesbezüglich von einer zu erwartenden nationalen Verordnung.
Der im Artikel genannte § 55(10) MDG ist in der verlinkten Version des Gesetzes nach meinem Kenntnisstand nicht (mehr) ganz aktuell und im aktuellsten Text des MPDG ist nur noch im §88 etwas ähnliches über eine Registrierungspflicht für Händler zu lesen die vier (mehr oder weniger spezifische) Händlergruppen betrifft. Könnten Sie dorthin gehend den Artikel (der mir in vielen Fällen schon sehr viel weitergeholfen hat(!)) aktualisieren?
Vielen Dank im Voraus!
Beste Grüße
Lukas de Hond
Sehr geehrter Herr de Hond,
eine nationale Durchführungsverordnung zur Regelung der Registrierungspflicht der Händler ist uns bisher nicht bekannt.
Die Möglichkeit zur Registrierungspflicht ist, wie Sie richtig schreiben, in §88 MPDG geregelt. Dort heißt es:
„9. festzulegen, dass Händler, die Produkte auf dem deutschen Markt bereitstellen, dies vor Aufnahme ihrer Tätigkeit bei der zuständigen Behörde anzuzeigen haben, sowie Inhalt und Form der Anzeige zu regeln.“
Den Beitrag haben wir entsprechend aktualisiert.
Freundliche Grüße
Luca Salvatore
Guten Tag,
weiter oben habe ich den Kommentar zum Artikel 13 (3) gelesen und das die Kennzeichnungspflicht am Produkt hängen würde. (Ich kopiere den Verlauf am Ende der Nachricht).
Diese Einschätzung irritiert mich nachhaltig. Der Artikel 13 beschreibt die Pflichten der Importeure und die Kennzeichnungspflicht obliegt nicht dem Hersteller, sondern dem Importeur. Wieso sollte dann der Artikel 120 Auswirkung auf die Verantwortung der Importeure haben?
Der Anhang I macht meines Wissens nach keine Angaben zur Kennzeichnung von Importeuren. Könnten Sie begründen, warum der Artikel 13 einen Zusammenhang zu MDD Produkten haben könnte.
Vielen Dank
Mit freundlichen Grüßen
Moritz Tillmann
„Sehr geehrter Herr Professor Johner,
soweit ich es verstanden haben gelten die Händler- und Importeurspflichten unabhängig von den Abverkaufsfristen bereits ab dem Geltungsbeginn der MDR am 26.05.21. Heißt dieses, dass wir z. B. in der Rolle als Importeur Ware, die wir nach diesem Termin erhalten, unabhängig davon, ob es sich um Legacy Produkte oder bereits nach MDR zertifizierte Produkte handelt, mit den Informationen zum Importeur kennzeichnen müssen oder bezieht sich dieses nur auf bereits MDR zertifizierte Produkte? Was ist mit bestehendem Lagerbestand, muss dieser auch gekennzeichnet werden?
Grüße
H. Simon
Luca Salvatore | Freitag, 5. März 2021 um 10:39 Uhr – Antworten
Sehr geehrter Herr Simon,
das ist eine spannende Frage. Nach unserem Verständnis hängen die Pflichten der Wirtschaftsakteure (Importeure/Bevollmächtigter/Händler) am Produkt. D.h., wenn ein Produkt noch nach MDD legal in Verkehr gebracht wird, muss derjenige erstmal „nur“ die MDD-Forderungen erfüllen, ausgenommen der Registrierungsanforderungen gemäß MDR/EUDAMED. Dies wurde uns von anwaltlicher Seite und auch durch ein Regierungspräsidium bestätigt. Eine Guidance oder ein Gerichtsurteil wären für diesen Fall dennoch sicherlich hilfreich, um eine definitive Aussage treffen zu können.
Herzliche Grüße
Luca Salvatore“
Sehr geehrter Herr Tillmann,
die von Ihnen referenzierte Antwort bezog sich allgemein auf die Frage, ob Artikel 13 auch für Importeure anwendbar ist, die weiterhin, nach dem 26.05.2021, ausschließlich Legacy-Produkte in den Verkehr bringen.
„Am Produkt hängen“ war vermutlich etwas missverständlich formuliert. Damit war gemeint, ob es sich um ein MDR-Produkt, MDD-Produkt (vor dem 26.05.2021 in Verkehr gebracht) oder Legacy-Produkt (Produkte, die nach dem 26.05.2021 weiterhin MDD-konform in Verkehr gebracht werden aufgrund von Übergangsbestimmungen) handelt. Dazu gibt es aktuell leider widersprüchliche Aussagen.
Freundliche Grüße
Luca Salvatore
Sehr geehrter Herr Professor Johner,
bei meiner Frage geht es um die Pflicht der Wirtschaftsakteure, die UDI der bezogenen und abgegebenen Produkte vorzugsweise elektronisch zu erfassen (Art.27,Abs.8), sofern es sich um Produkte der Klasse III handelt, oder sie zu den Produkten nach Absatz 11 lit. a gehören. Die Kommission legt die Produkte dafür fest.
Nun meine Fragen:
Gibt es bereits eine Auflistung von der Kommission?
Wo finde ich eine Liste dieser Produkte gem. Art. 27, Abs. 11,a?
Welche Produkte (welcher Klasse )sind hier enthalten?
Wir sind Hersteller der Produktklasse I. Sind unsere Kunden-vorzugsweise Sanitätshäuser- als Händler dann überhaupt verpflichtet, die UDI zu registrieren?
Vielen Dank im Voraus !
Mit freundlichen Grüßen
Christine Graß
Liebe Frau Graß,
mein Kollege Christopher Seib war so nett, die Frage zu beantworten:
Es gibt keine Auflistung, da es (noch) keine wie in Artikel 27 Absatz 11 erwähnten Durchführungsakten gibt. Also muss die UDI für Klasse-1-Produkte auch nicht erfasst werden.
Herzliche Grüße
Anja Segschneider | Redaktion
Sehr geehrte Frau Segschneider,
sehr geehrter Herr Seib,
vielen Dank für Ihre Antwort.
Ich habe dazu noch eine Nachfrage:
Wie verhält es sich dann mit Art. 25 (Identifizierung innerhalb der Lieferkette), insbesondere Art. 25, Abs. 2?
Geht es hier nur darum, Angaben über die beteiligten Wirtschaftsakteure machen zu können?
Oder muss zur Erfüllung dieser Anforderung der Händler- hier ein Sanitätshaus- explizit nachweisen können, welches Produkt von wem bezogen und an wen abgegeben wurde? Muss auch die Abgabe an den Endverbraucher(Patienten) dokumentiert werden?
Nochmal vielen Dank im Voraus!
Mit freundlichen Grüßen
Christine Graß
Liebe Frau Graß,
Wirtschaftsakteure nach MDR sind: Hersteller, Bevollmächtigte, Importeure, Händler und Artikel-22-Personen. Für diese gilt Artikel 25 (2) der MDR. D.h. der Händler muss angeben können (Nachverfolgbarkeit), welches Produkt er von welchem Wirtschaftsakteur erhalten hat (25 (2) b). Falls das Sanitätshaus ein Produkt an eine Gesundheitseinrichtung oder einen Angehörigen der Gesundheitsberufe abgibt, muss er auch dies einer Behörde nachvollziehbar zeigen können (25 (2) c)). Bei einer Privatperson (die kein Wirtschaftsakteur in diesem Sinne ist) muss keine Rückverfolgbarkeit gewährleistet sein.
Ich hoffe, das hilft Ihnen weiter!
Herzliche Grüße
Anja Segschneider | Redaktion
Sehr geehrte Frau Segschneider,
vielen Dank für Ihre Antwort. Sie haben mir damit sehr geholfen!
Mit freundlichen Grüßen
Christine Graß
Sehr geehrter Herr Prof. Johner,
Wäre es als Händler möglich durch eine CoC (Certificate of Conformity) pro Lieferung vom Hersteller, dem Artikel 14 genüge zu tun?
Mit freundlichen Grüßen,
Charlotte May
Liebe Frau May,
nein, das reicht leider nicht aus. Die MDR hat die Händlerpflichten als „Vier-Augen Prinzip“ gegenüber den Pflichten des Herstellers eingeführt. Von daher ist der Händler dazu verpflichtet den Artikel 14 selbständig umzusetzen.
Herzliche Grüße
Anja Segschneider
Sehr geehrter Herr Prof. Johner,
sehr geehrte Redaktion,
vielen Dank für Ihre tolle Website. Ich habe eine Frage zum Rückruf-Register welches der Händler führen soll:
Sollen hier Informationen aller Rückrufe des Produkts gesammelt werden oder nur Informationen über Rückrufe die auch Ware betrifft, welche der jeweilige Händler vertrieben hat?
Im ersten Fall müsste dann im Rahmen der QSV vereinbart werden, dass der Hersteller den Händler über jeglichen Rückruf informiert (auch wenn die betroffene Ware gar nicht an den Händler ging), oder?
Vielen Dank im Voraus
Guten Tag,
es handelt sich hierbei nur um Reklamationen und Rückrufe, bei denen Sie als Händler beteiligt waren. Sie müssen kein Verzeichnis über sonstige Rückrufe führen, an denen Sie nicht beteiligt waren.
Freundliche Grüße
Luca Salvatore
MPDG §88 gilt meiner Einschätzung nur und ausschließlich für Medizinprodukte bei deren Herstellung radioaktive Stoffe oder ionisierende Strahlen verwendet werden. Das sollte möglicherweise im Text klarifiziert werden.
Sehr geehrter Herr Schwarzar,
Besten Dank für Ihre präzise Anmerkung. Wir werden diesen Hinweis ergänzen.
Beste Grüße,
Pierre Jäger
Vor dem Hintergrund der zunehmenden Verbreitung von Software-Applikationen (kurz: Apps) die auch Eigenschaften eines Medizinproduktes aufweisen, hätte ich ein paar Fragen zur Regulierung, von denen ich annehme, dass sie auch von breiterem Interesse sind.
Vielleicht mögen Sie ja einmal versuchen, diese zu beantworten.
– Ein übliches Verfahren zur Distribution von Apps dürfte heute die Verteilung über einen AppStore sein.
D. h., der Hersteller einer App lädt diese auf einen App-Store hoch, von welchem sich der zukünftige Betreiber der App diese auf sein Endgerät herunterlädt.
Das muss gar nicht der PlayStore oder AppleStore sein, damit diese Technologie zum Einsatz kommt.
Die regulatorische Situation des Herstellers bei solchen Apps scheint mir noch einigermaßen übersichtlich, aber was ist eigentlich mit dem Betreiber des App-Stores, der ja i.d.R. von dem App-Hersteller verschieden sein wird?
Welche Rolle im Sinne eines Wirtschaftsakteurs (z. B. MDR) nimmt eigentlich der Betreiber des App-Stores ein? Ist das ein Händler?
Was ist, wenn der App-Store nicht in der EU gehostet wird, sondern in einem Drittstaat?
Ist der Betreiber des App-Stores dann Importeur?
Wer bringt dann eine App, die von dem App-Store heruntergeladen und auf einem Endgerät installiert wird in der EU Verkehr – und zu welchem Zeitpunkt geschieht das?
Beim Hochladen durch den Hersteller in den App-Store? Oder beim Herunterladen durch den Betreiber der App auf sein Endgerät?
Wer muss dementsprechend einen benannten Vertreter mit Sitz in der EU haben?
Der Hersteller? Oder der Betreiber des App-Stores? Oder beide?
– Für die Übergangsfristen der MDR macht es einen Unterschied, ob ein Medizinprodukt in Verkehr gebracht wird, oder ob ein bereits in Verkehr gebrachtes Medizinprodukt weiter auf dem Markt bereitgestellt wird.
Gibt es insofern bei über einen App-Store verteilten Apps einen Unterschied zwischen „Inverkehrbringen“ und „auf dem Markt bereitstellen“?
Was ist denn überhaupt für eine App der Zeitpunkt des Inverkehrbringens, bzw. wann beginnt und endet das „auf dem Markt bereitstellen“? Zum Zeitpunkt des Hochladens auf den App-Store, oder des Herunterladens durch den Betreiber der App auf sein Endgerät?
Es gibt vermutlich noch viele spannende Fragen, die mit dem klassischen Produktbegriff nur unzureichend beantworten sind.
Digitalisierung fängt im Kopf und bei der Gesetzgebung an, nicht erst bei der Beschaffung von Endgeräten und Glasfaserkabeln.
Sehr geehrter Herr Hagen,
das sind viele und spannende Fragen! Vielen Dank dafür! Ich teile Ihre Einschätzung, dass die Antworten viele Hersteller interessieren.
Daher habe ich diese Antworten (zumindest in Teilen) auf der Webseite zur Inverkehrbringung im 4. Kapitel adressiert. Wollen Sie prüfen, ob Ihnen die Antworten ausreichen? Falls nicht, dann haken Sie gerne nach. Wir tun unser Bestes, um eine Antwort zu liefern.
Mit den allerbesten Grüßen
Christian Johner
Sehr geehrter Herr Prof. Johner,
vielen Dank für die nützlichen Informationen. Bezüglich der Überprüfungspflicht der Händler, ob eine EU-Konformitätserklärung gem. Art. 14 (2) MDR ausgestellt wurde, fragen wir uns, wie Händler dieser Pflicht in der Praxis bis zur Funktionsfähigkeit von Eudamed nachkommen sollen, bzw. ob diese Pflicht bereits vor der Funktionsfähigkeit von Eudamed gilt? Wenn Händler keinen physischen Zugriff auf die Produkte selbst haben, müssen sie dann die Websites der Hersteller proaktiv und einzeln nach den Konformitätserklärungen absuchen bzw. ihre Dienstleister verpflichten, diese Kontrollen durchzuführen? Oder gibt es abgesehen von Eudamed derzeit auch eine Möglichkeit etwa in der DMIDS-Datenbank nach EU-Konformitätserklärungen zu suchen?
Vielen Dank für eine Antwort
MfG
Jennifer Ems
Sehr geehrte Frau Ems,
Besten Dank für Ihre spannenden Fragen. Es ist aktuell nicht klar wie der Händler dieser Prüfpflicht gerecht werden soll. Während der Ausarbeitung der Verordnung in initialer Fassung gab es wohl einen Vorschlag, den Hersteller zu verpflichten die Konformitätserklärung immer mitzuliefern. Letztendlich wurde dieser Vorschlag nicht aufgenommen. Wobei auch hier diskutiert werden kann, ob der Aufwand noch in einem akzeptablen Verhältnis zum Nutzen gestanden hätte.
Eines der jüngeren MDCG-Dokumente 2021-27 „Questions and Answers on Articles 13 & 14 of Regulation (EU) 2017/745 and Regulation (EU) 2017/746“ widmet sich ebenfalls dem Thema „Verification Obligations“ in Frage 15, jedoch liefert die Antwort wenig konkrete Hilfestellung. Letztendlich zeigt der letzte Halbsatz „[…] and otherwise, the manufacturer should be contacted.“, dass eine Kontaktaufnahme erwartet wird.
Für Sie als Händler ist die Situation nun in der Tat anspruchsvoll. Meine konkrete Empfehlung lautet:
1. Beschreiben Sie den Prüfvorgang in einer kleiner Verfahrensanweisung (VA) und dokumentieren Sie die Prüfungen in einer Checkliste.
2. Nutzen Sie die Möglichkeit des Stichprobenverfahrens, das Ihnen Art. 14 bietet explizit.
3. Beschreiben Sie die Vorgehensweise zum Prüfen der Konformitätserklärung in drei Stufen:
3a. Falls diese dem Produkt beiliegt –> ok / nicht ok
3b. Falls nicht ok, fragen Sie beim Hersteller oder Importeur oder Bevollmächtigten per E-Mail und ggf. Telefonanruf an. —> ok / nicht ok
3c. Falls nicht ok, suchen Sie auf der Webseite des Herstellers nach der Konformitätserklärung. –> ok / nicht ok
Falls 3c noch immer nicht zum Ziel führt, halten Sie in einem Kommentar fest, dass Sie diese Tätigkeiten umgesetzt haben und begründen Sie, warum Sie das Produkt trotzdem vertreiben.
Im DMIDS ist uns aktuell keine Möglichkeit bekannt nach Konformitätserklärungen zu suchen.
Auch wenn wir Ihnen keinen finalen Lösungsansatz liefern konnten, hoffen wir, dass unsere Rückmeldung dennoch hilfreich ist.
Beste Grüße
Pierre Jäger
Sehr geehrter Herr Jäger,
vielen Dank für die rasche Rückmeldung! Das hilft uns sehr weiter, vielen Dank!
Beste Grüße
Jennifer Ems
Sehr geehrter Herr Prof. Johner,
verstehe ich Ihre Ausführung zu den Händlerpflichten gemäß MDR Art 14 richtig, wonach eine „stichprobenartige“ Prüfung (u.a. CE-Kennzeichen vorhanden, Konformitätserklärung ausgestellt) durch das MPDG wieder ausgehebelt wird?
Also doch eine 100 %-WE -Prüfung und somit für jedes Medizinprodukt eine Konformitätserklärung beim Hersteller einholen?
Viele Grüße
Susanne
Liebe Frau Meyer,
herzlichen Dank für Ihre spannende Frage. Wir haben den Beitrag an der Stelle sowohl etwas geschärft, indem wir den Verweis auf den korrekten MPDG-Paragrafen eingefügt haben (§ 13 in Verbindung mit § 92 und nicht § 59), als auch etwas entschärft, indem wir die Interpretation zu den Fälschungen in direkten Bezug mit Art. 14 Abs. 2 Unterabsatz 3 setzen „Ist ein Händler der Auffassung oder hat er Grund zu der Annahme, dass ein Produkt nicht den Anforderungen dieser Verordnung entspricht […]“.
Der Händler hat unserer Einschätzung nach jedoch weder hieraus noch aus § 92 Abs. 1 Nr. 3 und § 13 MPDG die regulatorische Pflicht, sich über die stichprobenartigen Prüfungen hinaus auf etwaige Fälschungen zu prüfen, d.h. per Default misstrauisch zu sein und darauf basierend eine 100%-Prüfung festzulegen. Für die Strafvorschrift kommt es unserer Einschätzung nach auch darauf an, ob das unzulässige Anbieten oder Lagerhalten tatsächlich gefälschter Ware fahrlässig oder gar vorsätzlich erfolgte. Es wird somit kritisch, wenn der Händler weitere Anhaltspunkte oder Verdachtsmomente aufzeigt, dass er Fälschungen vertreibt.
In diesem Zusammenhang könnte das Stichprobenverfahren auch so ausgelegt werden, dass der Händler auch bestimmte „Anzeichen“ von Herstellern in Betracht zieht. Falls bei bestimmten Herstellern sich Fehler häufen, wie beispielsweise Fehler bei regulatorisch relevanten Dokumenten wie Konformitätserklärung, Gebrauchsanweisung oder auch der Anbringung der Kennzeichnung, oder ein Trend erkennbar ist, wäre durchaus eine Erhöhung der Proben empfehlenswert.
Ich hoffe, wir konnten Ihnen hiermit bereits helfen.
Herzliche Grüße
Pierre Jäger
Liebe Susanne,
Sie haben es vollkommen richtig vermutet: der Händler muss nur stichprobenartig die Konformitätsbewertung prüfen. Der Begriff Probenahmeverfahren bedeutet, dass die Größe der Stichprobe repräsentativ für die Menge der insgesamt verkauften Produkte für die die KOnformitätserklärung gilt, sein muss.
Mit herzlichen Grüßen
Astrid Schulze
Seniorberaterin QM & RA
Liebe Susanne,
im von Ihnen genannten Artikel heißt es weiter: „Der Händler hat unserer Einschätzung nach jedoch weder hieraus noch aus § 92 Abs. 1 Nr. 3 und § 13 MPDG die regulatorische Pflicht, sich über die stichprobenartigen Prüfungen hinaus auf etwaige Fälschungen zu prüfen, d.h. per Default misstrauisch zu sein und darauf basierend eine 100%-Prüfung festzulegen. “
Wir halten es also nicht für erforderlich, 100% zu prüfen. Es sei denn, der Händler hat auf Grund vorhergehender Prüfungen Grund zu der Annahme, dass eine Stichprobenprüfung nicht ausreicht. Bei Auffälligkeiten (z.B. bei mehrfach falschen Konformitätserklärungen) müsste die Stichprobe immer mehr vergrößert werden und kann im Extremfall bei der 100% Prüfung enden.
Mit herzlichen Grüßen
Astrid Schulze
Seniorberaterin QM & RA
Guten Tag Herr Johner,
ich habe eine Anmerkung bezüglich der MDR Artikel 16 (2) a und b.
Es geht um das kleine Wörtchen „erforderlich“ und welche Beziehungen es zu dem Thema Änderung der Verpackung, bereitzustellende Informationen usw. hat.
Hierzu gab es vor 2,5 Jahren (von Herrn Tillmann am 16. Oktober 2019) eine Nachfrage im Blog. Ihre Antwort lautete, dass das Wort „erforderlich“ sich aufgrund der Kommasetzung auf „was anderes“ bezieht und dieser Absatz generell den Händlern etwas mehr Möglichkeiten geben soll, da das OEM/PLM Geschäft endgültig hinfällig ist.
Wir haben das intern auch diskutiert und sind nach häufigem Lesen anderer Meinung. In der englischen Fassung ist im Übrigen der Satzbau auch dahingehend, eindeutiger meiner Meinung nach.
Des Weiteren existiert seit Oktober 2021 ein MDCG Papier (MDCG 2021-26) welches sich um genau diese Frage(n) kümmert. In diesem wird explizit das „Necessary in order to market“ erklärt und weiter auch nochmal darauf hingewiesen, dass die Notwendigkeit jedes Mal zu überprüfen ist „Whether a relabelling or repackaging activity is necessary, should be analysed on a case-by-case basis.“ Das finde ich, ist schon sehr deutlich wohin die Reise eigentlich geht!
Des Weiteren stehen dann Beispiele für solche Gründe (marketingtechnische für Händler sind nicht dabei).
Zusätzlich müssen laut Erklärung der Frage 5 den Herstellern und laut Frage 6 den zuständigen Behörden diese Gründe mitgeteilt werden!
Unserer Meinung nach werden dem Händler hier nur sehr bedingt Möglichkeiten geschaffen, sondern eher sehr stark reglementiert. Auch die Geschichte mit dem Zertifikat wird im MCDG 2021-23 erklärt und nun ist eigentlich klar, dass es ein „Produkt“-Zertifikat (z. B. bezogen auf einen MD-Code) ist und somit nicht durch die ISO 13485 (oder 9001) abgedeckt werden kann.
In Ihrem Artikel:
https://www.johner-institut.de/blog/regulatory-affairs/oem-original-equipment-manufacturer/
schreiben Sie:
„-Es sind mehrere Varianten dieser neuen Beziehung zwischen Hersteller (ex OEM) und Händler (ex PLM) denkbar:
-Der Hersteller liefert die Produkte als „White Label“ aus, der Händler übernimmt das Labeling.“
Unserer Meinung nach sind durch die zuvor angeführten Gründe die Erwartungen an das „White Label“ Produkt nicht, wie man es darunter verstehen könnte, gegeben, da es durch die Bedingung mit dem „‘Necessary in order to market“ extrem torpediert wird. Des Weiteren kann es eigentlich nicht als „White Label“ ausgeliefert werden, da es sich um ein bereits in Verkehr befindliches Produkt handeln muss… also vollends für den Endverbraucher gekennzeichnet bzw. verkehrsfähig. Das darf dann nicht geändert werden jedoch „bezusatzt“ werden.
Alles in allem führt es unserer Meinung dazu, dass das Konstrukt des „White Labels“ (wird in der MDR nirgends erwähnt, wurde aber durch den Artikel 16 irgendwie erwartet/impliziert), so überhaupt nicht möglich ist.
Entschuldigen Sie diese lange Ausführung, aber es ist ein komplexes Thema. Uns würde interessieren, welche Ansicht Sie diesbezüglich, auch unter Einbeziehung der Neuen MDCG-Papiere, mittlerweile vertreten
Vielen Dank und viele Grüße
Manfred Steinbach
Retec Kunststofftechnik GmbH
Lieber Herr Steinbach,
herzlichen Dank für Ihren ausführlichen Kommentar! Wie bereits per E-Mail erläutert prüfen wir die Anpassungen der Artikel basierend auf den neuen MDCG-Dokumenten.
Aufgrund der Detailtiefe können wir aktuell leider keine direkte Stellungnahme per Antwort in diesem Forum bereitstellen. Sie erhalten die Benachrichtigung sobald wir den Beitrag angepasst haben.
Herzliche Grüße
Pierre Jäger
Sehr geehrte Professor Johner,
wir sind uns nicht ganz im Klaren, ob wir das in Artikel 14(2), IVDR, genannte Probennahmeverfahren richtig interpretieren. Im Original lautet der Satz
„In order to meet the requirements referred to in points (a), (b) and (d) of the first subparagraph the distributor may
apply a sampling method that is representative of the devices supplied by that distributor.“
„representative of the devices supplied by that distributor“ könnte man unserer Meinung nach so auslegen, dass man eine Stichprobe aus allen gelieferten „Artikelnummern“ zieht. Beispiel: Wir handeln ein Portfolio von 100 Artikeln von Hersteller A und ein Portfolio von 200 Artikeln von Hersteller B. Pro Jahr überprüfen wir für 10 Artikel von Hersteller A und 20 Artikel von Hersteller B die geforderten Punkte. Im nächsten Jahr dann natürlich von 10 bzw. 20 anderen Artikeln dieser Hersteller.
Auf eine andere Frage zu Ihrem Blogbeitrag hat jedoch Ihre Kollegin Astrid Schulze mit einer anderen Interpretation des Probenahmeverfahrens geantwortet:
„Liebe Susanne,
Sie haben es vollkommen richtig vermutet: der Händler muss nur stichprobenartig die Konformitätsbewertung prüfen. Der Begriff Probenahmeverfahren bedeutet, dass die Größe der Stichprobe repräsentativ für die Menge der insgesamt verkauften Produkte für die die Konformitätserklärung gilt, sein muss.
Mit herzlichen Grüßen
Astrid Schulze
Seniorberaterin QM & RA“
Das liest sich so, als ob wir alle 100 Artikel von Hersteller A und alle 200 Artikel von Hersteller B prüfen müssten, aber eben nicht 100% der gelieferten Produkte, sondern nur eine gewisse Anzahl x. Das bedeutet natürlich dennoch einen erheblich größeren Aufwand, als nur eine Teilmenge des Portfolios überhaupt zu prüfen.
Könnten Sie hierzu noch einmal Klarheit schaffen? Das würde uns sehr weiter helfen.
Herzlichen Dank und viele Grüße,
Stefanie Schreyer
Liebe Frau Schreyer,
vielen Dank für die Frage!
Mein Kollege Pierre Jäger hat darauf die folgende Antwort gegeben:
„Eine bloße Orientierung an der Liefermenge ist mir etwas zu wenig und gibt auch dem Händler zu wenig Spielraum. Das wäre für mich – wie so oft – ein risikobasierter Ansatz, der in der Tat auch die Art des Produkts, die Risikoklasse, bisherige Beschwerden und Abweichungen berücksichtigt. Im Gesetzeswortlaut steht insofern nicht, dass das Verfahren für die verkaufte Menge (also im Sinne letztlich eines Prozentsatzes an Proben aus der Gesamtmenge) repräsentativ sein muss, sondern für das „von ihm gelieferte Produkt“ – was durchaus weiter geht. Umgekehrt erlaubt es der risikobasierte Ansatz, dass anfänglich größere Stichprobenmengen bei problemlosem Vertrieb reduziert werden, und so eine Entlastung des Händlers vorgenommen werden kann.
Beste Grüße
Pierre Jäger“
Sehr geehrter Herr Jäger,
ich habe folgende Fragen zum 3b und 3c aus Ihrem Beitrag vom Donnerstag, 6. Januar 2022:
– gibt es eine regulatorische/gesetzliche Verpflichtung für die Hersteller die Konformitätserklärung den Händlern zur Verfügung zu stellen? (z.B. nach Anfrage)
– gibt es eine Verpflichtung für die Hersteller die Konformitätserklärungen online zur Verfügung zu stellen?
Vielen Dank und viele Grüße,
Liebe Frau Anastasopoulos,
besten Dank für Ihre spannenden Fragen.
– gibt es eine regulatorische/gesetzliche Verpflichtung für die Hersteller die Konformitätserklärung den Händlern zur Verfügung zu stellen? (z.B. nach Anfrage)
Eine Verpflichtung gibt es für den Hersteller gemäß EU-MDR nicht direkt, jedoch ist ersichtlich, dass ein Händler seinen Verpflichtungen nicht nachkommen kann, wenn er diese nicht erhält. Deshalb sollte in der vertraglichen Regelung zwischen Hersteller und Händler festgehalten werden, dass die Konformitätserklärung bereitgestellt wird. Zudem fordert bspw. dass die EU-Konformitätserklärung für Produkte,
die auf dem deutschen Markt bereitgestellt werden, in deutscher oder in englischer Sprache zur Verfügung stehen muss (§ 8 Abs. 1 MPDG). Generell ist es schwer ersichtlich weshalb ein Hersteller dann noch die Herausgabe der Konformitätserklärung an den Händler verweigern kann.
– gibt es eine Verpflichtung für die Hersteller die Konformitätserklärungen online zur Verfügung zu stellen?
Eine solche Vorgabe ist uns nicht bekannt.
Weitere Regelungen auf europäischer Ebene sind wünschenswert und wir hoffen, dass diese bald nachgeführt werden. Ich hoffe dennoch, dass Ihnen unsere Antwort bereits weiterhilft.
Herzliche Grüße
Pierre Jäger
Liebes Johner Team,
Vielen Dank für den übersichtlichen Artikel.
Ich habe eine Frage zur Kennzeichnung.
Ein Medizinprodukt soll in Deutschland und in der Schweiz auf den Markt gebracht werden. Der Hersteller sitzt in der EU und ist auch entsprechend auf dem Produkt angegeben. Es sollen die selben Packmittel für Deutschland und die Schweiz verwendet werden. Ist es für den Vertrieb in Deutschland ein Problem, dass der Ch- Rep und der Ch- Importeur auf der Packung angegeben ist? Es gibt zusätzlich einen Händler in Deutschland der das Produkt direkt vom Hersteller kauft. Dieser ist aber nicht auf der Packung angegeben. Das ist doch, solange er keine Tätigkeit entsprechend Artikel 16 der MDR durchführt, auch nicht gefordert oder?
Vielen Dank für ihre Mühe.
Beste Grüsse
Liebe Frau Fränkle,
Die Angaben von mehreren Bevollmächtigten auf der Verpackung wäre zulässig. Die Bevollmächtigten sind ja mit den entsprechenden Labels CH-REP etc. gekennzeichnet.
Liebe Grüsse, Mario Klessascheck
Sehr geerhtes Johner Team,
ich habe eine Frage bezüglich Weiterverkauf von Medizinprodukten durch Händler, bzw. Hersteller, bei zugekauften Produkten.
Muss ein Händler, bzw. Hersteller, wenn er bereits zugelassene Medizinprodukte anderer Hersteller verkauft gem. MDR sicherstellen, dass die IFU, sowie das Label mit Artikelbezeichnung in der Landesspache des Bestimmungslandes, beigefügt wird?
Vielen Dank für Ihre Antwort.
Simon Beck
Sehr geehrter Herr Beck,
Besten Dank für Ihre Fragen. Generell obliegt die Verantwortung in erster Linie dem Hersteller, wie unter anderem Artikel 10 Absatz 11 und Anhang II Kapitel 2. erster und zweiter Spiegelstrich fordern. Jedoch erkennt man an den Anforderungen aus Artikel 14, der explizit für Händler anwendbar ist, Paragraph 2b), dass der Händler eben genau gemäß dem zuvor genannten Artikel prüfen nämlich: „b) dem Produkt liegen die vom Hersteller gemäß Artikel 10 Absatz 11 bereitgestellten Informationen bei;“.
Artikel 10 Absatz 11 lautet im Detail „(11) Die Hersteller sorgen dafür, dass dem Produkt die Informationen gemäß Anhang I Abschnitt 23 in einer oder mehreren von dem Mitgliedstaat, in dem das Produkt dem Anwender oder Patienten zur Verfügung gestellt wird, festgelegten Amtssprache(n) der Union beiliegen. Die Angaben auf der Kennzeichnung müssen unauslöschlich, gut lesbar und für den vorgesehenen Anwender oder Patienten klar verständlich sein.“
Ich hoffe, wir können Ihnen damit helfen.
Beste Grüße
Pierre Jäger
Hallo liebes Johner-Team,
könnten Sie Artikel 16 (1) a) näher eingehen?
Im folgenden Fall:
Der Händler wünscht sich ein Medizinprodukt, auf dem sein Name/Marke steht. Das ist meiner Meinung nach laut der MDR nicht mehr möglich, da der Händler damit genauso Hersteller wäre und die Pflichten an einen Hersteller erfüllen muss. In diesem Fall wären wir nur ein ausgelagerter Prozess und unser Händler ist der Hersteller und Händler gleichermaßen.
Jedoch verwirrt mich der Artikel 16 (1) a) sehr, da es so klingt, als ob dies doch möglich wäre. Grundvoraussetzung dafür wäre dann eine Vereinbarung unsererseits? Oder müssten wir das Medizinprodukt unter unserem Namen inverkehrbringen und der Händler labelt dies dann auf seinen Namen um?
Siehe auch 1.d) hier im Blog: „Natürlich wäre es auch denkbar, dass der Hersteller das Produkt im „Design“ des Händlers in den Verkehr bringt. Der Hersteller muss aber immer (auch) auf dem Label erkennbar sein. Das fordert Artikel 16, Absatz 3.“
Würde mich freuen, etwas mehr Licht ins Dunkle zu bekommen. 🙂
Liebe Grüße
Liebe Frau Häuslmeir,
das Konstrukt ist weiterhin möglich und wird beschrieben als Own-brand labelling oder Virtual Manufacturing. Wichtig ist dabei insbesondere, dass der Hersteller, der das Produkt Inverkehrbringen wird, auf dem Produkt als solcher kenntlich ist, dass er über die vollständige Technische Dokumentation verfügt (anders als bei der früheren PLM/OEM-Verbindung mit Part A / Part B) und dass ein Vertrag geschlossen wird wie in Art. 16 (1) gefordert.
Falls Sie das Konstrukt im Detail beleuchten möchten können wir dies gerne im Rahmen eines Beratungsmandats anbieten.
Ich hoffe, dass Ihnen diese Rückmeldung hilft.
Beste Grüße
Pierre Jäger
Liebes Johner-Team,
Vielen Dank für Ihre tolle Seite und den hilfreichen Informationen. Dennoch habe ich eine Frage.
Der Mutterkonzern sitzt innerhalb der EU und verpackt dort Arzneimittel mit Medizinprodukten (Behandlungseinheit z.B. Fertigspritze mit Butterfly, Alkoholtupfer etc.) und gibt diese frei. Anschließend werden diese Behandlungseinheiten über das Tochterunternehmen in Deutschland in Verkehr gebracht. Muss das Tochterunternehmen die geforderten Prüfungen als Händler durchführen? Da die Medizinprodukte (z.B. Spritzen, Tupfer etc.) in der Arzneimittelverpackung integriert sind, müssten für die stichprobenartige Prüfung mehrere Packungen somit zerstört und vernichtet werden. Kann man dies umgehen und sich auf die Prüfung des Mutterkonzerns verlassen (z.B. durch Verträge)? Wie ist in diesem Fall die MDR anzuwenden?
Vielen Dank für Ihre Hilfe.
Beste Grüße
Lieber Herr Mertens,
Besten Dank für Ihren Kommentar zu unserem Händler-Artikel.
Wenn Ich Ihrer Beschreibung korrekt folge, wäre es durchaus möglich, dass der der Mutterkonzern, der die Produkte in der EU Inverkehrbringen wird, die Prüfung selbst so aufzeichnet, dass alle relevanten Anforderungen aus Art. 14 abgedeckt sind.
Wir kennen dieses Vorgehen auch von mehreren größeren Unternehmen, die für ihre Händler (häufig auch Töchter bzw. verknüpfte Firmen) die Prüfungen durchführen und über einen Fileshare die Unterlagen bereitstellen. Das alles wäre dann auch vertraglich zu regeln. Gerade bei einer Mutter-Tochter-Beziehung ist es hier durchaus möglich die Erfüllung der Anforderungen an die Situation entsprechend anzupassen.
Beste Grüße
Pierre Jäger
Sehr geehrter Herr Jäger,
ich habe eine Frage zum Artikel 16 1a,MDR.
Wenn eine Niederlassung (die die Händlerrolle inne hat) den Produktnamen ändert, wird sie dann zum Hersteller?
Dieser neue Produktname weicht von der Produktbezeichnung in der Konformitätsbewertung ab.
Ist diese Umbenennung und somit Abweichung überhaupt zulässig?
Vielen Dank für Ihre Hilfe.
Mit freundlichen Grüßen
C.Graß
Sehr geehrte Frau Graß,
besten Dank für Ihre Fragen!
Den Produktnamen darf der Händler nicht ändern. Ausnahme wäre natürlich, wenn dieser einen entsprechenden Vertrag mit dem Hersteller hat, und somit beispielsweise im Modell Own-brand labelling / Virtual Manufacturing agiert, was gemäß Art. 16 1a (zweiter Teil) ermöglicht wird.
Beste Grüße
Pierre Jäger
Liebes Johner-Team,
gemäß MDR Artikel 14 (4) : „(…) Die Händler arbeiten mit dem Hersteller (…) zusammen, um sicherzustellen, dass bei Bedarf die erforderlichen Korrekturmaßnahmen ergriffen werden, um die Konformität des Produkts herzustellen, es vom Markt zu nehmen oder zurückzurufen.“
Was bedeutet dies genau für den Händler im Rückruffall (Sicherheitsmaßnahme vom Hersteller)?
– muss der Händler seine Kunden selbst informieren? die Produkte selbst zurückholen? oder reicht es, wenn er die Kundendaten an dem Hersteller weiterleitet?
– wer trägt eventuelle Kosten für den Rückruf (z.B. Transport- und Bearbeitungskosten)?
Vielen Dank im Voraus für Ihre Rückmeldung.
Viele Grüße
C. Anastasopoulos
Liebe Frau Anastasopoulos,
Besten Dank für Ihre spannenden Fragen!
Deshalb lautet unsere Empfehlung auf Verträge zwischen Hersteller und Händler hinzuarbeiten, um genau solche Punkte, die das Innenverhältnis betreffen, zu regeln.
Hierzu empfehlen wir Herstellern unter anderem auch sogenannte Mock-Recalls durchzuführen, um die Belastbarkeit der Ketten zu testen.
Bei Pflichtverletzungen des Händlers kann es durchaus sein, dass der Hersteller / Importeur oder weiterer zuliefernder Händler Regressanforderungen stellt, um Transport-/Bearbeitungskosten beim nächst gelegenen Wirtschaftsakteur zu platzieren, z.B. wenn Händler Ihren Prüfpflichten gemäß Art. 14 nicht nachgekommen sind. In erster Instanz steht jedoch der Hersteller im Fokus und muss diese Kosten entsprechend einplanen, was üblicherweise auch in entsprechenden Produkthaftpflichtpolicen berücksichtigt wird.
Beste Grüße
Pierre Jäger
Liebes Johner-Team,
Vielen Dank für Ihre tolle Seite und den hilfreichen Informationen. Sie haben mir schon sehr weitergeholfen.
Ich habe eine Frage zu Medizinprodukteakten, die ich als Händler nach Artikel 14 archivieren muss ?
Wir handeln mit Produkten der Klassen I und II. Unser Sortiment umfasst ca. 3000 verschiedene Artikel.
Bei einem Audit teilte uns der Auditor mit, dass wir Medizinprodukteakten der gehandelten Produkte vorweisen müssten.
Da ich neu im Handel bin und vorher bei einem Medizinproduktehersteller gearbeitet habe, bei dem ich selbst Medizinprodukteakten der Klasse III erstellt habe, war ich etwas überrascht. Ich konnte mir nicht vorstellen, wie ein Händler an die vollständigen Akten der Hersteller gelangen soll. Er sagte, es gehe z.B. um Konformitätserklärungen, IFUs, Sicherheitsdatenblätter etc.
Die Überprüfung auf richtige Kennzeichnung, ob Konformitätserklärungen vorliegen und IFUs den Produkten beiliegen, machen wir stichprobernartig.
Müssen die IFUs und Konformitätserklärungen zu den Produkten archiviert werden und welche Dokumente müssen noch archiviert werden?
Vielen Dank im Voraus für Ihre Rückmeldung.
Viele Grüße
Simon
Lieber Simon,
besten Dank für Ihre positive Rückmeldung und Ihre Fragen.
Sie sollten sich bei der Frage nach der Archivierung am Besten entlang Ihrer Pflichten bewegen, d.h.
– Konformitätserklärung
– CE-Kennzeichnung (mit / ohne Benannte Stelle und korrekte Nummer)
– Gebrauchsanweisung
– UDI (gemäß Fristen)
– Vorkommnisse
– Bestellungen / Lieferungen
– entsprechende Begründungen für Ihr Stichprobenverfahren
– Ausgefüllte Prüfdokumente zu den zu überprüfenden Dokumenten
Bei den Fristen der Archivierung richten Sie sich bitte nach Art. 10(8) der MDR/IVDR d.h. 10 Jahre / 15 Jahre (Implantate).
Was Sie über den Auditor berichten kann ich in diesem Kontext leider nicht beantworten. Möglicherweise haben Sie Anforderungen in Ihrem QM-System definiert, die in diese Richtung deuten.
Möglicherweise wäre es eine Option, dass einer unserer Berater:innen ein Probeaudit mit Fokus auf Ihre Pflichten durchführt, um danach für das nächste Audit besser gerüstet zu sein. Was meinen Sie dazu?
Melden Sie sich gerne bei uns bspw. über das Kontaktformular.
Beste Grüße
Pierre Jäger
Liebes Johner-Team,
vielen Dank für diesen wirklich sehr interessanten und informativen Beitrag. Meine Frage schließt sich an die letzte Frage von Simon an und betrifft die Erfassungs- und Archivierungspflichten des Händlers.
Wir beziehen von einem österreichischen Hersteller Medizinprodukte der Klasse Ir (wiederverwendbare chirurgische Instrumente) und verkaufen diese in Deutschland. Nach Art. 123 Abs. 3 lit. f) MDR gilt die Kennzeichnungspflicht mit einem UDI-Träger für die Risikoklasse I erst ab dem 26. Mai 2025. Der Hersteller hat der Produktverpackung aber bereits eine UDI angebracht. Diese beinhaltet GTIN=UDI-DI, LOT Nummer (Teil der UDI-PI) und Herstellungsdatum (Teil der UDI-PI).
Unsere Beschaffung arbeitet jedoch (noch) nicht mit GTIN. Muss dies (also die ganze UDI) in unser Warenwirtschaftssystem aus rechtlicher Sicht übernommen werden oder reicht die LOT Nummer (für die spätere Rückverfolgbarkeit) aus? Wenn wir die GTIN grundsätzlich übernehmen müssen, haben wir eine Übergangszeit (also müssen wir es erst ab dem 26.05.2025 zwingend machen oder schon früher, weil der Hersteller bereits jetzt eine UDI anbringt)?
Ich selbst habe dazu den Art. 27 Abs. 8 MDR gefunden, wonach die Wirtschaftsakteure (also auch wir als Händler) die UDI der Produkte, die sie abgegeben oder bezogen haben, nur (vorzugsweise elektronisch) erfassen und speichern müssen, sofern diese Produkte zu Folgendem gehören:
— den implantierbaren Produkten der Klasse III;
— den Produkten, Produktkategorien oder Produktgruppen, die von einer der in Absatz 11 Buchstabe a genannten Maßnahmen erfasst werden.
Ich verstehe es so, dass wir die UDI nicht zwingend erfassen oder speichern müssen, weil die oben genannten Voraussetzungen in unserem Fall nicht erfüllt sind. Reicht es also bei uns grundsätzlich für das angemessene Niveau der Rückverfolgbarkeit i.S.d. Art. 25 Abs. 1 MDR aus, wenn wir auch künftig, also auch nach dem 26.05.2025 nur die LOT-Nummer erfassen und archivieren?
Ich bin ganz neu in dieser Thematik und freue mich daher über jede Hilfe.
Vielen Dank schon im Voraus für Ihre Rückmeldung!
Viele Grüße
Heleri
.
Liebe Frau Heleri,
besten Dank für Ihre spannenden Fragen!
Ich lese zwei Fragen heraus, die ich nachfolgend nochmals formuliere:
1. Ab wann müssen UDI verwendet werden?
Hier haben Sie vollkommen recht und beziehen sich auf die noch nicht erreichte Frist. Sofern ich Ihre Produktangaben korrekt verstehe, gilt für diese – gemäß Art. 123 (3) g) iii) sogar eine noch längere Frist zur Anbringung, nämlich der Mai 2027.
2. Müssen wir wirklich die UDI speichern / verarbeiten?
Sie haben auch dort bereits den richtigen Artikel bzw. das richtige Kapitel der MDR gefunden.
a) Sie müssen gemäß Art. 25 ein „angemessenes Niveau der Rückverfolgbarkeit von Produkten“ erreichen. Dazu ist die geschilderte Verfolgung mittels LOT in Ordnung. Und dementsprechend auch die Archivierung.
b) die unter Art. 27 (8) genannten Anforderungen sind für Sie nicht anwendbar.
Sie sollten natürlich in Ihren internen Vorgaben den ARt. 14 (2) d) der MDR in diesem Zusammenhang prüfen d.h. „… UDI vergeben?“ und das gemäß Ihres Stichprobenplans prüfen. Aktuell kommen Sie bei den zuvor genannten Produkten zum Ergebnis „Anbringung noch nicht gefordert“.
Beste Grüße
Pierre Jäger
Sehr geehrtes Johner Team,
Szenario: Händler (A) bezieht vom Hersteller (B) einen Rollator.
Der Kunde informiert Händler (A), dass die Fußstützen gebrochen sind.
Der Händler (A) informiert den Hersteller (B) und erhält neue Fußstützen zur Reparatur.
Händler (A) tauscht die defekten Fußstützen aus und verpackt den Rollator wieder.
1. Greifen bei der Aufbereitung bzw. Reparatur von Medizinprodukten die Art. 16 (1) c und/oder Art. 16 (2) b der MDR für den Händler, so dass dieser möglicherweise sogar Hersteller wird?
2. Muss der Händler (A) den Prozess des Verpackens in diesem Fall zertifizieren lassen?
Sehr geehrter Herr Wilde,
Besten Dank für Ihre Nachricht und die spannenden Fragen.
Ich gehe davon aus, dass Sie die Erlaubnis / Befähigung haben diese Reparaturen und erneutes Verpacken durchzuführen, was absolut üblich ist.
zu 1. Nein, die zitierten Abschnitte greifen in diesem Fall nicht, denn sie bringen das Produkt wieder zurück in den Einklang mit dessen Spezifikation. Und sie verändern das Produkt auch nicht, um es in einem bestimmten Land vermarkten zu können, sondern, wie zuvor geschrieben, bringen es zurück in Übereinstimmung mit der Spezifikation. Der Verpackungsprozess ist zudem sehr gut nachvollziehbar.
zu 2. Eine Zertifizierung ist aus meiner Sicht nicht notwendig. Wie zuvor beschrieben ist es sinnvoll und insbesondere auch im Interesse des Herstellers eine vertragliche Regelung zu etablieren, sodass die beschriebenen Tätigkeiten dort geregelt sind.
Beste Grüße
Pierre Jäger
Sehr geehrtes Johner Team,
Vielen Dank für die Ausführung zum Thema „Anforderungen an die Händler“.
Ich habe eine Frage bezüglich der Pflichten der Händler.
Muss ein Händler (EU) gegenüber dem Hersteller (EU) offenlegen in welche weiteren EU-Länder er die Produkte verkaufen möchte?
Ich denke hierbei an die vom Hersteller bereitzustellenden Informationen, wie die Gebrauchsanweisung in der jeweiligen Landessprache – welche dann nicht immer gewährleistet ist.
Ist es zulässig dies in einem Distribution Agreement festzuhalten?
Viele Grüße,
Steffi
Liebe Steffi,
Ihr solltet das auf jeden Fall im Vertrag regeln. Und ja, es kann sein, dass der Hersteller das offengelegt haben möchte. Wir haben das beispielsweise auch schon bei sogenannten Mock-Recalls angefragt, um die Erfüllung der Anforderungen zu überprüfen, damit wir im Falle eines Recalls möglichst effektiv und effizient agieren können. So interpretiere ich auch Art. 25 (1) der 2017/745 „Die Händler und Importeure arbeiten mit den Herstellern oder ihren Bevollmächtigten zusammen, um ein angemessenes Niveau der Rückverfolgbarkeit von Produkten zu erreichen.“
Und auch die Erfüllung der spezifischen Anforderungen zu Sprachen muss erfüllt, damit der Hersteller seine Pflichten nach Art. 10 (11) und der Händler seine Prüfpflichten nach Art. 14 (2) b) erfüllen kann.
Beste Grüße
Pierre Jäger
Sehr geehrtes Johner Team,
mein Frage wäre, benötigt man als Ausländisches Medizintechnikunternehmen der ein CE Zeichen nach MDR haben möchte, eine Niederlassung oder ein Händler in der EU?
Und welcher § regelt dies?
Sehr geehrter Herr Zrinski,
besten Dank für Ihre Meldung und die spannenden Fragen.
Es hängt davon ab, wo der Sitz des „ausländischen Unternehmens“ ist. Falls es sich um ein sogenanntes Drittland handelt, dann benötigt der Hersteller einen europäischen Bevollmächtigten gemäß MDR / IVDR Art. 11, der als regulatorischer Repräsentant agiert und auf dem Produkt angegeben werden muss. Zudem gibt es in den meisten Fällen einen europäischen Importeur gemäß MDR / IVDR Art. 13, der gekennzeichnet werden muss.
Das Johner Institut bzw. dessen Tochterfirmen können diese Dienstleistung in Europa sowie der Schweiz und UK anbieten.
Beste Grüße
Pierre Jäger
Hallo –
Ich würde gerne Ihre Meinung zu SaMD (konkret eine Webseite, die in der EU gehostet wird) einholen und wer dabei als Distributor auftritt.
Haben gerade den Fall, dass eine Behörde gem. nationalen Gesetz eine Registrierung eines Distributors verlangt.
Vielen Dank,
Beste Grüße
Lieber Peter M.,
Besten Dank für Ihre Nachricht.
Gemäß Ihrer Schilderung kann ich leider nicht den gesamten Kontext erschließen, weshalb eine passende Antwort sehr schwer ist. Ein Link zum betroffenen Medizinprodukt (SaMD) bzw. der Webseite wäre sehr hilfreich. Ich interpretiere es so, dass die Webseite als SaMD qualifiziert ist, sofern ich Ihren Kommentar richtig verstehe.
Beste Grüße
Pierre Jäger
Hallo Pierre,
korrekte Einschätzung. Die Website berechnet anhand von 5 Parametern einen möglichen Therapieoutcome.
Hilft das weiter?
Danke
VG Peter
Lieber Peter M.,
danke für Ihre Reaktion. Um die Situation vollständig bewerten zu können, wäre die vollständige Vernetzung zwischen Hersteller und Händler zu betrachten, was jedoch die Einschätzung per Antwort auf einen Fachartikel nicht zulassen würde.
Generell gibt es regionale Pflichten zur Registrierung von Händlern in einigen Ländern. Falls der Hersteller die Webseite hostet und der Händler diese bewirbt wäre erkennbar, dass jener als Händler auftritt.
Falls Sie eine tiefergehende Analyse benötigen können Sie sich gerne per Kontaktformular an unsere Experten wenden, die anschließend bspw. ein Angebot für ein Fachgutachten erstellen. Dafür würden wir mit einer Geheimhaltungsvereinbarung sicherstellen, dass Sie uns alle relevanten Informationen teilen können.
Beste Grüße
Pierre Jäger
Guten Tag,
danke für den wertvollen Beitrag!
Bei den Fulfillment centern bin ich noch unsicher: sind diese Anbieter tatsächlich „Händler“ im Sinne der MDR? QSVs mit Verkaufsriesen wie Amazon erscheinen mit praktisch nicht durchzubringen.
Ich freue mich auf Ihr Feedback.
Einen angenehmen Arbeitstag noch!
Hallo,
Vlt. ein anregender Literaturbeitrag
https://doi.org/10.1038/s41746-023-00754-6
VG
Peter M.
Danke für Ihre wertvollen Ergänzungen. Gemäß unserer Interpretation passen diese Unternehmen sehr gut in die Kategorie „Fulfilment-Dienstleister“ gemäß der Verordnung über Marktüberwachung und die Konformität von Produkten https://eur-lex.europa.eu/legal-content/DE/TXT/PDF/?uri=CELEX:32019R1020
Leider wird dieser Wirtschaftsakteur nicht in der Medical Device Regulation 2017/745 genannt. Und die Anwendung der zuvor genannten Verordnung größtenteils ausgeschlossen für Medizinprodukte.
Deshalb sind wir an dieser Stelle leider ebenfalls nicht sicher wie die Einordnung dieser Unternehmen erfolgt. Wir hoffen auf entsprechende Klarstellungen oder gar Gerichtsurteile.
Beste Grüße
Pierre Jäger
Liebes Johner-Team,
wie wird es im Rahmen der Verlängerung der Übergangsfristen für Händler erkennbar sein, ob MDD-Zertifikate länger gültig sind? Werden neue ausgestellt? Oder wird z.B. eine Erklärung des Herstellers, dass die Bedingungen erfüllt sind, reichen?
Vielen Dank im Voraus für Ihre Rückmeldung,
viele Grüße,
Liebe Frau Anastasopoulos,
der Händler wird sich dies vom Hersteller versichern lassen können bzw. müssen. Kommen berechtigte Zweifel auf, wird der Händler das Produkt nicht anbieten dürfen und muss den Hersteller konfrontieren. Dieses Vorgehen entspricht dem Grundsatz des Art. 14 Abs. 2 MDR.
Beste Grüße
Pierre Jäger
Sehr geehrtes Johner Team,
zunächst danke für den Artikel und dem wertvollen Verlauf der darauffolgenden Kommentar- und Antwortserie. Ich selbst ringe im Augenblick mit Artikel 16 (2) und ob dieser in Bezug auf den Absatz 1 zu verstehen ist. Stehen die Aufgaben aus Artikel 16 (2) a) und b) in direktem Bezug zu Artikel 16 (1), oder müssen diese womöglich losgelöst vom Vertrieb unter eigenem Handelsnamen betrachtet werden? Konkret lautet meine Frage, müssen die Forderungen nach (3) und (4) auch dann umgesetzt werden, falls die ursprüngliche Kennzeichnung der Handelsmarke, des Herstellers unberührt bleiben? Ein konkretes Beispiel wäre ein vom Hersteller ausgelagerter Prozess an den Importeur/Händler zur Umverpackung/Kennzeichnung, der das Ergänzen des Produktes durch das Beilegen einer Gebrauchsanweisung in der jeweiligen Amtssprache, unter gleichzeitiger Beibehaltung des Handelsnamens/Marke und der Wirtschaftsakteure regelt. Freundliche Grüße!
Lieber Boris E.
um die komplexen Anforderungen gemäß Art. 16 Abs. 3 und 4 MDR zu vermeiden, ist es möglich (wie in Ihrem Beispiel beschrieben), die Tätigkeiten Umpacken und Übersetzen der IFU als ausgegliederten Prozess des Herstellers zu gestalten, so dass der Importeur/Händler diese Tätigkeiten im Auftrag und unter Verantwortung des Herstellers durchführt. Denn Art. 16 Abs. 3 und 4 MDR stellt darauf ab, dass Importeure oder Händler diese Tätigkeiten eigenverantwortlich durchführen (typischerweise im Bereich des Parallelimports).
Die Tätigkeiten als ausgegliederter Prozess sollten in einer QSV mit dem Hersteller nachvollziehbar dokumentiert werden.
Beste Grüße
Pierre Jäger
Sehr geehrtes Johner Team,
folgende Fragestellung begleitet uns derzeit. Wir kaufen Medizinprodukte (speziell für die Wundversorgung) innerhalb der EU ein. Entsprechend ist das Produkt bereits mit einem CE Kennzeichen versehen und verfügt über eine Konformitätserklärung.
Auf dem Produkt (Sekundärpackmittel) etikettieren wir unsere eigene PZN und geben unseren Firmennamen sowie unsere Anschrift bekannt. Das Produkt wird ansonsten nicht weiter verändert oder geöffnet. Zudem übersetzen wir auch keine Texte und handeln lediglich Produkte mit deutscher Beschriftung inkl. Deutscher Packungsbeilage.
Demnach dürften wir lt. MDR nicht unter Artikel 16 fallen sondern werden unter Artikel 14 als Händler klassifiziert? Ist meine Annahme richtig?
Entsprechend besteht dann keine Verpflichtung, dass unser QMS System von einer unabhängigen Stelle zertifiziert wird?
Vielen Dank für Ihre Unterstützung
Guten Tag,
ich sehe in Ihrem Fall ebenfalls Artikel 16 (2) als nicht anwendbar, da Sie die äußere Verpackung nicht wesentlich ändern, z.b. durch Umverpackung oder Übersetzung der Kennzeichnung. Solange Sie also die originäre Kennzeichnung durch die zusätzliche Angabe der PZN nicht beeinträchtigen (z.b. durch Überkleben von Anwenderinformationen), wäre in Ihrem Fall nur Artikel 14 anwendbar.
Freundliche Grüße
Luca Salvatore
Liebes Johner-Team,
dürfen wir als Händler denn Medizinprodukte der Klassen I und IIa in den Verkehr bringen, welche ein Symbol auf den Hinweis einer Bedienungsanleitung auf dem Label haben, die Anleitung aber nicht physisch in der Verpackung ist, sondern nur online als eiFU zur Verfügung steht, dieses aber noch nicht auf dem Label angepasst ist?
Wie lange hat der der Hersteller Zeit, das Label dementsprechend anzupassen, wenn nur diese eIFU existiert?
Oder ist das alles im Ermessensbereich, wenn der Hersteller diese eIFU auch auf Anfrage zur Verfügung steht und man diese Anleitung dem Kunden, mit seiner Einwilligung weiterleitet bzw. der Kunde sogar auf die Anleitung verzichtet?
Mit den besten Wünschen
Lieber Herr Ganz,
besten Dank für Ihre Nachricht und die spannenden Fragen.
Der Hersteller muss die Durchführungsverordnung für eIfU (2021/2226) beachten und eine Risikobewertung durchführen. Wir gehen davon aus, dass der Hersteller dies getan hat.
In dieser Verordnung ist unter anderem geregelt, dass der Hersteller – auf Anfrage – eine gedruckte Form der Gebrauchsanweisung innerhalb einer Frist bereitstellen muss. Meine Empfehlung wäre hierfür eine simulierte Anfrage durchzuführen.
Was die Kennzeichnung betrifft ist diese – formal gesehen – nicht korrekt und müsste angepasst werden. Es müsste der Hinweis auf eIfU erscheinen.
Soweit die formale Sichtweise.
Risikobasiert argumentiert ist es ggf. möglich, dass der Hersteller eine Bewertung der Abweichung durchführt und zur Schlussfolgerung kommen könnte, dass er das Label innerhalb einer gesetzten Frist ändert und bis dahin das Produkt nicht vollständig konform mit den Vorgaben ist. Dies sollte auf Hersteller- wie auf Händlerseite schriftlich festgehalten werden. Die Frist sollte nachvollziehbar sein, und der Hersteller könnte sich bspw. an Hill/Schmitt, Medizinprodukterecht, 20. Lfg., Art. 12 MDR Rn. 2 orientieren, welcher zwar auf die Änderung des Bevollmächtigten abzielt, jedoch scheint eine Frist von 6 Monaten (maximal) als angemessen.
Beste Grüße
Pierre Jäger
Vielen Dank Herr Jäger,
das Hauptproblem von Regulatory Affairs ist, dass auf dem Label nicht der Hinweis auf eIFU geben ist.
Das hat mir schon mal weiter geholfen, so kann ich jeden Lieferant damit konfrontieren.
Leider ist es so, dass viele Hersteller erst nach der erneuten MDR-Zulassung des Produktes ihre Label umgestalten bzw. nach MDR anpassen.
Mit den besten Wünschen
Michael Ganz
Arthrex GmbH
Sehr geehrtes Team des Johner Institut,
wir sind als Händler für sehr viele Hersteller in Europa ansässig und versenden Europaweit Medical Devices. Hierbei ist die „good distribution practice“ (GDP) nicht verpflichtend. Gibt es im Geltungsbereich der MDR eine Verpflichtung, eine Transportvalidierung durchzuführen oder wie könnte man möglicherweise abbilden, dass man die Umgebungsbedingungen der Produkte bei einem Transport Europaweit berücksichtigt hat. Ist man ebenso verpflichtet, einen GDP zertifizierten Transporteur zu beauftragen, oder kann man hier „jedes x-beliebige“ Transportunternehmen nehmen? MFG Paul Beuth
Lieber Herr Beuth,
vielen Dank für Ihre spannende Frage!
Die MDR fordert gemäß Artikel 14(3), dass Händler für die Einhaltung der Lagerungs- und Transportbedingungen gemäß den Vorgaben der Hersteller verantwortlich sind, während sich die Produkte in der Verantwortung der Händler befinden. Es gibt keine explizite Anforderung an Händler, eine Transportvalidierung durchzuführen. Dies liegt in der Verantwortung der Hersteller. Abhängig von den durch den Hersteller definierten Umgebungsbedingungen für das Produkt, sollte die Lagerung und der Transport durch die Händler ggf. überwacht und kontrolliert erfolgen. Beispielsweise kann eine Temperaturverteilungsmessung im Transportfahrzeug als Nachweis zur Einhaltung der vorgesehenen Temperaturgrenzen dienen. Es gibt eine ganze Reihe an Logistikunternehmen, die spezielle Anforderungen an die Umgebung gewährleisten können. Eine Abweichung von den Herstellervorgaben während der Lagerung und des Transports durch den Händler kann dazu führen, dass dieser für mögliche resultierende Produktmängel verantwortlich ist.
Sehr geehrte Johner Team
uns ist unklar welche Vorkommnisse welcher Bundesbehörden gemeldet werden müssen.
Es ist Klar, dass schwerwiegende Vorkommnisse den Zuständigen Behörden gemeldet werden müssen.
jedoch was muss dem BFARM und was muss dem Pei gemeldet werden,
muss man als IVD Händler der Klasse C und D dann diese Produkte zusätzlich noch bei einem Vorkommnis an dem Pei melden. also an das BFARM UND PEI am Ende ?
Hängt also die Meldung an den Bundesbehörden von der Klassifizierung der Produkte ab.
wenn es dann Tatsächlich einen Rückruf gibt, der vom Hersteller aus angefordert wird , sollten wir dies dann dem Pei melden oder dem BFARM. oder Beide
Mit freundliche Grüßen,
Fernando
Lieber Fernando,
vielen Dank für Ihre spannende Frage.
Ich stimme Ihnen zu, es ist in der Tat auf den ersten Blick nicht ganz eindeutig geregelt. Doch die Zuständigkeit des PEI beschränkt sich auf bestimmte IVDs. Demzufolge ist das PEI Für alle IVDs der Klasse D und einen Teil der Klasse C (gemäß Regel 3 Buchstabe a bis e und g des Anhang VIII der Verordnung (EU) 2017/746) die zuständige Bundesoberbehörde für die zentrale Erfassung und Bewertung von Vorkommnis-Meldungen gemäß Artikel 82 IVDR.
Unter folgendem Link finden Sie die notwendigen Informationen: https://www.pei.de/DE/arzneimittelsicherheit/ivd-vigilanz/ivd-vigilanz-node.html.
Für diese Produkte wäre demnach die Meldung beim PEI ausreichend.
Sehr geehrtes Johner-Team,
wir sind Hersteller von Medizinprodukten, haben also ein 13485-Zertifikat, sind aber auch Händler für Vertriebsprodukte. Wir ändern nichts, fallen also nicht unter den Artikel 16. Wir haben aber den Vertrieb von „Einmalartikeln“ noch mal als Extra-Satz auf unserem Zertifikat stehen. Jetzt haben wir ein Vertriebsprodukt im Werden-Prozess, dessen Lagerbedingungen wir im Haus nicht einhalten können. Mit dem Artikel und dem Blog bin ich mir jetzt noch nicht ganz sicher, wie das mit der Implementierung eines Außenlagers für ein Vertriebsprodukt ist. Sind wir als Händler in der Verantwortung das neue Lager unserer Beannten Stelle zu melden (wenn dort nicht unsere eigenen Produkte lagern)? Müssen wir das neue Lager implementieren wie eines für unsere eigenen Produkte, also die Benannte Stelle miteinbeziehen? Oder würde nur der Hersteller dieser Produkte zur Verantwortung gezogen werden als insgesamt „ausgelagerten Prozess“, was oben im Blog zu einem anderen Thema bereits erwähnt wurde? Würde also theoretisch eine Freigabe des Herstellers für dieses Außenlager genügen?
Vielen Dank im Voraus
Nicole
Liebe Nicole,
die MDR fordert im Artikel 14(3), dass Händler von Medizinprodukten die Lagerungs- und Transportbedingungen gemäß den Vorgaben des Herstellers sicher stellen müssen. Die Verantwortung dafür verbleibt also bei Ihnen als Händler. Dazu gehört unter anderem, dass Sie ein entsprechendes (ausgelagertes) Lager qualifizieren und überwachen. Die Nachweise dieser Tätigkeiten wird der Hersteller von Ihnen anfordern bzw. die Einhaltung der spezifischen Lager- und Transportbedingungen für seine Produkte ggf. sogar bei Ihnen selbst auditieren. Händler haften bei einem Verstoß gegen die definierten Lager- und Transportbedingungen. Die Anforderungen und Verpflichtungen dahingehend regeln Sie am besten vertraglich mit dem Hersteller.
Das entsprechende Lager müsste in Ihrem Fall nur dann Ihrer Benannten Stelle angezeigt werden, wenn dadurch mögliche Tätigkeiten im Rahmen einer Bescheinigung gemäß Artikel 16(4) oder auch die Lagerung ihrer eigenen Medizinprodukte, bei denen Sie als Hersteller auftreten, betroffen wären.
Herzliche Grüße
Manuela Reinhold
Sehr geehrtes Johner-Team,
Wir kaufen ein Beleuchtungssystem für unser IVD-Mikroskope bei Firma A (außerhalb der EU). Firma A hat auch Importeure in der EU. Wir möchten den Namen des Beleuchtungssystems ändern und unser Logo darauf anbringen und auf dem Typenschild lassen wir den Namen des Herstellers.
Das Beleuchtungssystem ändern wir technisch nicht, wir passen es an unsere Software an, damit das Beleuchtungssystem mit unserer Software gesteuert werden kann.
Dazu habe ich 2 Fragen:
1. Können wir unseren Firmennamen auf dem Gerät haben und auf dem Typenschild den ursprünglichen Hersteller A stehen lassen, so dass wir nicht Hersteller sind?
2. Wie können wir den Namen ändern, ohne Hersteller zu sein?
Mit der Namensänderung ist auch die Firma A gemeint.
Vielen Dank
Sehr geehrter Almuaz,
bitte entschuldigen Sie die aufgrund der Feiertage und Urlaubszeit verspätete Rückmeldung.
Die Beantwortung Ihrer Frage hängt sehr stark von der generellen Handhabung des Beleuchtungssystem ab. Stellen Sie das Beleuchtungssystem vom Hersteller A als Zubehör zu Ihrem Produkt bereit, dann treten Sie als Händler auf und dürfen den Namen des Produkts nur dann ändern, wenn dies gemäß Artikel 16(1) mit dem Hersteller vereinbart wurde und der Hersteller auch weiterhin als solcher auf der Kennzeichnung ersichtlich bleibt. Gibt es hingegen keine Vereinbarung, befindet sich Ihr Name auf dem Gerät und der Name von Firma A weiterhin auf dem Beleuchtungssystem. Möchten Sie als Händler für das Beleuchtungssystem in Erscheinung treten, dürfen Sie Ihren Namen und Anschrift z.B. durch ein zusätzliches Etikett auf dem Produkt anbringen ohne die originäre Kennzeichnung des Herstellers zu beeinträchtigen.
Verbauen Sie das Beleuchtungssystem hingegen als Komponente in Ihr Medizinprodukt, dann befindet sich das Gesamtprodukt in Ihrer Verantwortung und Sie sind als Hersteller auf dem Gesamtprodukt erkennbar. Das Typenschild der Firma A kann in diesem Fall entfernt werden.
Sehr geehrtes Johner-Team,
in unserer Rolle als Händler von Medizinprodukten verpacken wir manche unserer Handelswaren um (Transportverpackung). Beispielsweise bekommen wir vom Hersteller 12 Verkaufseinheiten (VE) in einer Transportverpackung, wir verkaufen sie jedoch als einzelne VE, d.h. sie werden in einer anderen (mit anderen Abmaßen) Transportverpackung an unsere Kunden geliefert. Müssten wir in diesem Zusammenhang eine Transportvaldierung der kleineren Transportverpackung durchführen?
Vielen Dank im Voraus
Anett
Liebe Anett,
vielen Dank für die spannende Frage.
Gemäß dem Leitfaden MDCG 2021-26 dürfen Sie als Händler die Transportverpackung ändern, um die Produkte in kleineren Versandeinheiten auf dem Markt bereit zu stellen. Die äußere Verpackung (inkl. Kennzeichnung) darf dabei nicht beeinträchtigt werden. Die explizite Forderung nach einer Transportvalidierung gibt es meines Wissens nach nicht. Ich empfehle Ihnen, sich hinsichtlich der Wahl der Verpackungsart (Kartonage, Kunststoffbehälter, usw.) an der ursprünglichen Transportverpackung des Herstellers zu orientieren. Entscheiden Sie risikobasiert, ob die Produkte bzw. deren äußere Verpackung durch den Transport beeinträchtigt werden könnten. Ggf. können Sie sich auch bei dem Hersteller rückversichern, dass der Versand in kleineren Transportverpackungen über die Transportvalidierung des Herstellers abgedeckt ist. Bitte beachten Sie in diesem Zusammenhang, dass Sie gemäß Artikel 14 (3) der MDR für die Einhaltung der Lagerungs- und Transportbedingungen verantwortlich sind, wenn sich die Produkte in ihrer Verantwortung befinden. Wenn Sie mit Ihrer gewählten Transportverpackung also keine spezifischen Vorgaben des Herstellers missachten, steht einer Änderung der Transportverpackung zur Vereinzelung nichts entgegen.
Sehr geehrtes Johner-Team,
vielen Dank für den informativen Artikel. Sie haben oben u.a. ja beschrieben, dass es für die Hersteller sinnvoll ist, mit den Händlern QSV’s abzuschließen. Meine Frage hierzu wäre: ist ein Händler nicht automatisch auch ein „Lieferant“? Denn er bietet ja letztendlich eine Dienstleistung an, die ich „outsource“. Somit kämen dann automatisch die Pflichten der 13485:2016 Punkt 7.4.1 Beschaffungsprozess (Stichwort: risikobasierte Lieferantenauswahl) für mich als Hersteller zum Tragen und auch eine QSV nach 4.1.5 müsste sowieso erstellt werden. Oder wäre damit der Begriff des Lieferanten zu streng gefasst? Ich müsste dann ja den Händler auch regelmäßig bewerten und überwachen?
Herzlichen Dank und mit freundlichen Grüßen
Kai Just
Lieber Herr Just,
vielen Dank für Ihre spannende Frage. Grundsätzlich ist es eher untypisch, einen Händler gleichzeitig als Lieferant im QM-System zu führen. Gemäß ISO 9000 ist ein Lieferant eine „Organisation, die ein Produkt oder eine Dienstleistung bereitstellt.“ Als Beispiele listet sie dabei Hersteller, Vertriebseinrichtungen, Einzelhändler oder Verkäufer eines Produkt oder einer Dienstleistung. Die Definition würde demzufolge streng genommen zwar anwendbar sein, allerdings passt die Verkaufsrichtung nicht. Das würde bedeuten, dass der Händler den Hersteller als Lieferant listen müsste.
Ein Händler wäre nur dann als Lieferant zu führen, wenn ein Hersteller einen Prozess gezielt an einen Händler auslagert, z.B. die Übersetzung einer Gebrauchsanweisung in eine andere Sprache. In diesem Fall sollten die Anforderungen an die Qualität der Übersetzung in einer QSV geregelt werden. Hier kommt auch der von Ihnen referenzierte Abschnitt 7.4.1 der ISO 13485 zum Tragen.
Beste Grüße
Manuela Reinhold
Hallo liebes Johner-Team,
wir kaufen Medizinprodukte zu (sind selbst nicht Hersteller) und verkaufen diese an unseren Händler in Belgien. Unser belgischer Händler vertreibt diese in Belgien.
Müssen wir uns in Belgien in der FAMHP als Händler registrieren, auch wenn wir in Belgien selbst nicht die Medizinprodukte vertreiben?
Vielen Dank im Voraus!
Schöne Grüße,
Maximilian
Lieber Herr Fürhauser,
die landesspezifischen Registrierungsanforderungen der EU-Mitgliedstaaten sind in den entsprechenden nationalen Gesetzen enthalten. In Belgien müssen sich gemäß dem Royal Decree vom 15. November 2017 alle Händler mit Sitz in Belgien und auch Händler, die auf belgischem Territorium tätig sind, ihre Tätigkeit im FAMHP Web Portal registrieren. Wenn Sie Medizinprodukte an einen belgischen Händler verkaufen, stellen Sie per Definition diese Medizinprodukte auf dem belgischen Markt bereit. Meiner Einschätzung nach fallen Sie damit unter die Registrierungspflicht in Belgien. Die FAMHP hat als Entscheidungshilfe auch einen übersichtlichen Flowchart bereit gestellt: https://www.famhp.be/sites/default/files/content/POST/MEDDEV/10%20web%20portail/Flow%20chart%20distributeur%20EN.pdf
Melden Sie sich gerne bei uns, wenn wir Sie mit der Recherche nach lokalen, landesspezifischen Anforderungen unterstützen können.
Herzliche Grüße
Manuela Reinhold
Für Medizinprodukte welche außerhalb der EU hergestellt werden und in der EU vertrieben werden benötigt man auf dem MD ja die Angabe des Herstellers und des EC Rep. Kann in diesem Fall auch der Ec Rep der Importeur sein? Laut MDR muss ja auch der Importeur auf der Kennzeichnung der Verpackung des MD angegeben sein oder sehe ich das falsch?
Lieber Herr Hopf,
damit liegen Sie richtig. Medizinpropdukte, die außerhalb der europäischen Union hergestellt werden, benötigen neben dem Bevollmächtigten (EC Rep) einen Importeur, der die Produkte innerhalb der Union in Verkehr bringt. Der Bevollmächtigte und der Importeur können dieselbe natürliche oder juristische Person mit Sitz in der Union sein. Die Pflichten eines Importeurs sind im Artikel 13 der MDR festgelegt. Dazu gehört auch eine Kennzeichnungspflicht gemäß Artikel 13(3), d.h. der Importeur muss auf dem Produkt oder auf seiner Verpackung oder auf einem dem Produkt beiliegenden Dokument seinen Namen sowie die Anschrift angeben. Ich empfehle Ihnen dahingehend auch unseren Blogbeitrag zu den Importeuren, der die Rolle und deren Pflichten nochmals schön zusammenfasst: https://www.johner-institut.de/blog/regulatory-affairs/importeure/.
Herzliche Grüße,
Manuela Reinhold
Guten Tag,
bin ich als Händler eines Medizinprodukts (Produkt wird nicht unter meinem Namen vertrieben) verpflichtet, ein Qualitätsmanagementsystem zu haben ? Das Produkt soll auf dem deutschen Markt verkauft werden, der Hersteller, unter dessen Namen das Produkt vertrieben wird, befindet sich in Frankreich.
Vielen Dank für eine kurze Antwort.
Liebe Frau Wolthusen,
Händler von Medizinprodukten, die keine Tätigkeiten nach Artikel 16 (2) ausüben, müssen kein (zertifiziertes) Qualitätsmanagementsystem vorhalten. Grundsätzlich empfehlen wir Händlern jedoch immer ein Qualitätsmanagementsystem aufzubauen. Der Umfang sollte dabei zumindest die Pflichten eines Händlers gemäß Artikel 14 der MDR abdecken. Auf diesem Weg kann sicher gestellt werden, dass der Händler z.B. seinen Prüfpflichten nachkommt und ein Register der Beschwerden, der nichtkonformen Produkte und der Rückrufe und Rücknahmen führt.
Händler, die jedoch Tätigkeiten im Sinne des Artikel 16 (2) ausüben, müssen hingegen ein QM-System etablieren, dass den Vorgaben des MDCG 2021-23 entspricht und durch eine Benannte Stelle gemäß Artikel 16 (4) zertifiziert wird.
Herzliche Grüße,
Manuela Reinhold
Sehr geehrtes Johner-Instituts-Team!
Folgendes Thema: Wir sind Medizinproduktehersteller in Europa und auch Händler bzw. Importeur. Folgendes Beispiel – wir kaufen Medizinprodukt A (Klasse IIa) – dieses wird im Säckchen zu je 40 Stück geliefert. Wir wollen diese in kleineren Mengen weiterverkaufen (z.B. 10er Säckchen) und müssen daher umverpacken, Damit fallen wir unter Artikel 16 2(b). Ich wollte fragen, was das für Auswirkungen auf die UDI hat? Die UDI die wir vom Kunden erhalten haben bezieht sich meines Verständnisses ja auf die Packungsgröße 40 Stück und nicht auf kleinere Mengen. Ich würde daher sagen, man bräuchte eine neue UDI – ich als Umverpacker darf aber doch keine eigene UDI anbringen? Oder soll ich die UDI vom Hersteller so übernehmen wie ich sie bekomme, ohne dabei zu berücksichtigen, dass es bei uns kleinere Verkaufseinheiten sind?
Ich freue mich auf Ihre Antwort.
Mit lieben Grüßen,
Kathrin
Liebe Frau Steiner,
Ihre Frage ist absolut berechtigt. Der Umgang mit der UDI unter Anwendung von Artikel 16 (2) ist nicht näher geregelt, sogar eher missverständlich geregelt. Fakt ist, dass nur der Hersteller die UDI vergibt, nicht der Händler. Um die Frage vollumfänglich zu beantworten, müssten wir dafür Ihren Fall näher prüfen. Wenn Sie dabei Unterstützung benötigen, dann melden Sie sich gerne bei uns.
Herzliche Grüße,
Manuela Reinhold
Sehr geehrtes Johner-Instituts-Team, bei einem externen Audit wurde durch die Benannte Stelle gefordert, dass ein Handelsunternehmen bei der Stichprobenprüfung nach Art. 14 MDR bzw. IVDR sicherstellt, dass die vorliegenden Produkte rechtskonform in Verkehr gebracht wurden, insbesondere wenn sie unter die Übergangsregelungen der Verordnung (EU) 2023/607 fallen.
Konkret geht es um Produkte, die ursprünglich auf Basis einer abgelaufenen Konformitätserklärung gemäß MDD bzw. IVDD in Verkehr gebracht wurden, aber aufgrund der verlängerten Übergangsfristen weiterhin verkehrsfähig sind – vorausgesetzt, der Hersteller hat rechtzeitig einen Vertrag über eine Konformitätsbewertung nach MDR bzw. IVDR mit einer Benannten Stelle abgeschlossen.
Sind Händler verpflichtet, im Rahmen der Stichprobenprüfung ein Bestätigungsschreiben („Confirmation Letter“) der Benannten Stelle beim Hersteller anzufordern, welches das Bestehen eines entsprechenden Vertrags mit dem Hersteller belegt?
Falls ja, müssen die betroffenen Produkte bis zur Vorlage eines solchen Nachweises durch den Hersteller für den weiteren Vertrieb gesperrt werden, um der Sorgfaltspflicht nachzukommen?
Vielen Dank für Ihre Einschätzung
Liebe Frau Petzold,
vielen Dank für die spannende Frage. Weder die MDR noch die MDCG regeln diese Prüfpflicht nochmal weiterführend im Detail. Mit den verlängerten Übergangsfristen haben wir aktuell auch eine erschwerte Situation, die sich erst löst, wenn der Übergang zur MDR vollständig geschafft ist. Ich teile gerne unsere Ansicht mit Ihnen: um die Prüfpflicht gemäß Artikel 14(2) a) mit entsprechender Sorgfalt erfüllen zu können, ist es äußerst ratsam zu prüfen, ob die Produkte unter die verlängerten Übergangsfristen gemäß Artikel 120 MDR bzw. 110 IVDR fallen und trotz abgelaufener Zertifikate in Verkehr gebracht werden dürfen. Hersteller können entweder über eine Selbsterklärung bestätigen, dass sie die Bedingungen an diese Übergangsfristen erfüllen oder weisen dies über einen Confirmation Letter ihrer zuständigen Benannten Stelle nach. Mindestens eins dieser Dokumente sollten Sie zusammen mit den Zertifikaten bzw. den Konformitätserklärungen beim Hersteller anfordern, um bewerten zu können, ob Sie als Händler diese Produkte weiter auf dem Markt bereit stellen dürfen. Der letzte Absatz im Artikel 14(2) untersagt Ihnen auch die weitere Bereitstellung auf dem Markt, wenn das Produkt nicht konform zur MDR ist. Wenn der Hersteller alle Bedingungen erfüllt, sollte es theoretisch für ihn kein Problem sein, eine der beiden Erklärungen vorweisen zu können.
Ich hoffe, das hilft Ihnen weiter.
Herzliche Grüße,
Manuela Reinhold
Vielen herzlichen Dank! Ihre Antwort bestätigt meine Vermutung.
Liebes Johner-Team,
Als Händler von Medizinprodukten müssen wir ja auch überprüfen, ob der Hersteller eine UDI vergeben hat. Ist das offensichtliche Vorhandensein der UDI mit Angabe der UDI-DI, Lot-Nummer und Ablaufdatum inkl. (Data Matrix) Code auf dem Produkt/der Verpackung ausreichend oder gehört die Überprüfung des maschinenlesbaren Codes ebenfalls dazu? Konkret geht es um die Kennzeichnung eines Klasse I-Produkts mit GS1, bei der zwar die von Menschen lesbaren Angaben korrekt sind, allerdings die GS1-Data Matrix nach Scan mittels des GS1 Healthcare Barcode Scanners nicht den GS1-Standards entspricht. Dürfen wir das Produkt dann überhaupt bereitstellen bzw. ist es dann unsere Pflicht, den Hersteller darauf hinzuweisen?
Vielen Dank vorab!
Liebe Frau Kirschenhofer,
wenn man dem Wortlaut der MDR folgt, so ist gemäß Artikel 14 (2) unter Punkt d) lediglich die Rede davon zu prüfen, ob der Hersteller eine UDI vergeben hat. Wie bei vielen anderen Punkten auch, ist es bislang nicht vollends geklärt, was der Händler konkret prüfen muss. Theoretisch könnte diese Prüfung auch mit Hilfe der Konformitätserklärung erfolgen, insofern der Hersteller die UDI auf der Konformitätserklärung auch angibt. Doch eine bloße Prüfung der Vergabe einer UDI ist meiner Ansicht nach nicht wirklich sinnig, wenn nicht auch die verschlüsselten Angaben geprüft werden, vor allem unter Berücksichtigung der Compliance-Verantwortung eines Händlers innerhalb der Vertriebskette. Denn letztendlich soll ein Händler ja auch Produktfälschungen und nicht-konforme Produkte identifizieren und diese dann auch entsprechend melden können. Ich empfehle also, um der durch die MDR auferlegten Compliance-Verantwortung der Händler gerecht zu werden, eine Prüfung des maschinenlesbaren Teils der UDI durchzuführen und den Hersteller auf die Nicht-Konformität hinzuweisen. Die MDR fordert im Anhang VI unter Punkt 4.11, dass der UDI-Träger bei normaler Anwendung während der erwarteten Lebensdauer des Produkts lesbar sein muss. Ist er das nicht, liegt eine Nichtkonformität vor und der Hersteller ist verantwortlich. Letztendlich liegt die Prüftiefe aber in Ihrem Ermessen, bis die EU-Kommission oder die MDCG weiterführende Informationen dazu veröffentlicht.
Ich hoffe, das hilft Ihnen weiter.
Herzliche Grüße,
Manuela Reinhold
Hallo Dr. Manuela Reinhold,
vielen Dank für ihre interessanten Artikel. Ich habe neben dem Artikel „Medizinprodukt als Handelsware“ auch den „Anforderungen an Händler (die auch die Hersteller betreffen)“ gelesen und habe in diesem Kontext Fragen zum Umgang mit wiederverwendbaren chirurgischen Instrumenten:
Wenn man als Händler im Rahmen der Aufbereitung notwendige Reparaturen an den Instrumenten vornimmt, bleibt man dann in seiner Rolle aus Händler oder wird man rechtlich unter Umständen zum Hersteller? Und wenn ja, unter welchen Umständen? Und wenn nein, brauche ich dann als Händler trotzdem ein (zertifiziertes) QMS?
Mit freundlichen Grüßen
Kris
Lieber Herr Wegener,
vielen Dank für Ihre spannende Frage, die sich leider nicht pauschal beantworten lässt. Ob der Händler zum Hersteller wird, wenn er Reparaturen an wiederverwendbaren chirurgischen Instrumenten vornimmt, hängt stark von den Umständen ab. Insofern der Hersteller Wartungs- und Reparaturvorgaben in seiner Gebrauchsanweisung macht, können diese Arbeiten gemäß den Angaben des Herstellers durchgeführt werden. Werden Reparaturen ohne das Vorhandensein solcher Vorgaben des Herstellers von einem Händler (eigenständig) vorgenommen, ist das eine Tätigkeit, die einen Einfluss auf die Konformität des Produktes haben kann. Damit läuft der Händler Gefahr, selbst zum Hersteller zu werden, auf den die Pflichten eines Herstellers gemäß Artikel 10 der MDR dann vollständig übergehen.
Melden Sie sich gerne bei uns, wenn Sie Bedarf haben, Ihre Fragen tiefergehend mit uns zu besprechen. Wir freuen uns darauf.
Herzliche Grüße
Manuela Reinhold