
Die chemische Charakterisierung nach ISO 10993-18 ist ein zentraler Bestandteil der Biokompatibilitätsbewertung gemäß ISO 10993-1 und damit eine Voraussetzung für die Zulassung von Medizinprodukten.
Sie dient dazu, unbekannte Substanzen in Medizinprodukten zu identifizieren, um eine toxikologische Risikobewertung durchführen zu können.
Dieser Artikel verschafft einen Überblick über
- die chemische Charakterisierung und deren Analysemethoden,
- die gesetzlichen Anforderungen an Medizinproduktehersteller,
- typische Fehler und Best Practices, um schnelle Zulassungen und sichere Produkte zu erreichen.
- FDA-konforme Analysen
- Hilfestellung bei problematischen Testergebnissen
- Toxikologische Risikobewertung
- Regulatorische Unterstützung beim Q-Sub Meeting oder bei Rückfragen Benannter Stellen
- Pragmatische und lösungsorientierte Bewertung der Biokompatibilität
- Strategische Planung – schlank und konform
1. Chemische Charakterisierung gemäß ISO 10993-18
a. Definition
Chemische Charakterisierung: Prozess des Erhalts von chemischen Informationen, entweder durch Informationserfassung oder durch Informationsermittlung, z. B. durch Literaturrecherche oder chemische Prüfung
Chemische Information: qualitative und gegebenenfalls quantitative Kenntnis über die Konfiguration, Zusammensetzung und Herstellung des Medizinprodukts und/oder seiner Herstellungswerkstoffe, anhand deren die Identitäten und Mengen der in den Werkstoffen und im Medizinprodukt enthaltenen Bestandteile festgestellt werden
b. Chemische Charakterisierung als Prozess
Die chemische Charakterisierung nach ISO 10993-18 ist ein Prozess zur Sicherstellung von chemischen Informationen für die biologische Bewertung von Medizinprodukten. Grundsätzlich lassen sich hier drei Prozessarten unterscheiden:
- Nachweis der chemischen Äquivalenz: Vergleich eines neuen Medizinprodukts mit einem bereits zugelassenen Produkt zur Feststellung der Gleichwertigkeit
- Abgleich mit Werkstoffnormen: Überprüfung, ob die chemische Zusammensetzung eines Produkts mit einschlägigen Normen und regulatorischen Vorgaben übereinstimmt
- Grundlage für die toxikologische Risikobewertung nach ISO 10993-17: Identifizierung und Quantifizierung von Bestandteilen eines Medizinprodukts zur Bewertung der Biokompatibilität gemäß ISO 10993-1
Beachten Sie weiterführende Informationen zur toxikologischen Risikobewertung nach ISO 10993-17.
2. Rolle der Datenblätter bei der chemischen Charakterisierung
a. Welche Datenblätter es gibt
Sicherheitsdatenblätter (SDS) – Arbeitsschutz und Gefahrenbewertung
Sicherheitsdatenblätter (SDS) liefern wichtige Informationen, z. B. zur Zusammensetzung, sicheren Handhabung, Lagerung und Entsorgung von Chemikalien und Gemischen. Rohstofflieferanten, Hersteller oder Vertreiber sind gesetzlich verpflichtet, ein SDS bereitzustellen für
- Gefährliche Chemikalien: wenn die Substanz oder ein Gemisch oder einzelne Inhaltsstoffe als gefährlich eingestuft werden (z. B. reizend, toxisch, umweltgefährlich)
- PBT oder vPvB-Stoffe: Stoffe, die als persistent, bioakkumulierbar und toxisch (PBT) oder sehr persistent und sehr bioakkumulierbar toxisch (vPvB) eingestuft sind und damit ein Potenzial zur Anreicherung in einem Organismus haben
- Besonders besorgniserregende Stoffe: Stoffe auf der Kandidatenliste für besonders besorgniserregende Stoffe gemäß Artikel 59.1 der REACH-Verordnung, z. B. CMR
Die Angabe klassifizierter Inhaltsstoffe basiert auf regulatorischen Vorgaben wie REACH (EG 1907/2006) bzw. CLP-Verordnung (EG 1272/2008) in der EU sowie dem Globally Harmonized System (GHS) weltweit. Entsprechend klassifizierte Stoffe, die mindestens 0,1 % des Gesamtgewichts ausmachen, müssen deklariert werden.
Technische Datenblätter (TDS) – Materialeigenschaften und Anwendungsdaten
Technische Datenblätter (TDS) enthalten detaillierte Material- und Anwendungsspezifikationen, darunter:
- Physikalische, mechanische und thermische Eigenschaften (z. B. Temperaturbeständigkeit, Dichte, Härte, Elastizität)
- Chemische Beständigkeit (z. B. Resistenz gegenüber Chemikalien und Lösungsmitteln)
- Zusammensetzung, Reinheit und Qualität des Materials oder Produkts
- Normen und Zertifizierungen, Anwendungsbeispiele (Medizinprodukte)
Ein TDS ist nicht gesetzlich vorgeschrieben. Es wird aber von vielen Herstellern bereitgestellt, um Produkthaftungsrisiken zu minimieren und Informationen zu vermitteln.
Zertifikate – Regulatorische Compliance
Zertifikate werden als Bescheinigung der Einhaltung regulatorischer Vorgaben ausgestellt.
- Biokompatibel nach ISO 10993-Reihe oder USP Class VI
- Food grade
- Weitere
b. Weshalb Datenblätter nicht ausreichen
Obwohl SDS und TDS sowie Zertifikate wertvolle chemische Informationen liefern, reichen sie für eine vollständige chemische Charakterisierung nach ISO 10993-18 nicht aus. Die Gründe:
- Unvollständige Angaben zur chemischen Formulierung: Hersteller von Rohstoffen, insbesondere Polymere, geben oft keine detaillierten Identitäten und Mengenangaben.
- Keine Informationen zu freisetzbaren Substanzen: Die chemische Zusammensetzung allein sagt nichts darüber aus, welche Substanzen unter klinischen Bedingungen tatsächlich aus dem Medizinprodukt im finalen Zustand austreten.
- Abhängigkeit von der Verwendung der Materialien: Die Art der Verarbeitung der Materialien, in der Produktion eingesetzte Hilfsstoffe und die Alterung bzw. Reaktion der Materialien mit anderen wirken sich darauf auf, welche Stoffe freigesetzt werden können.
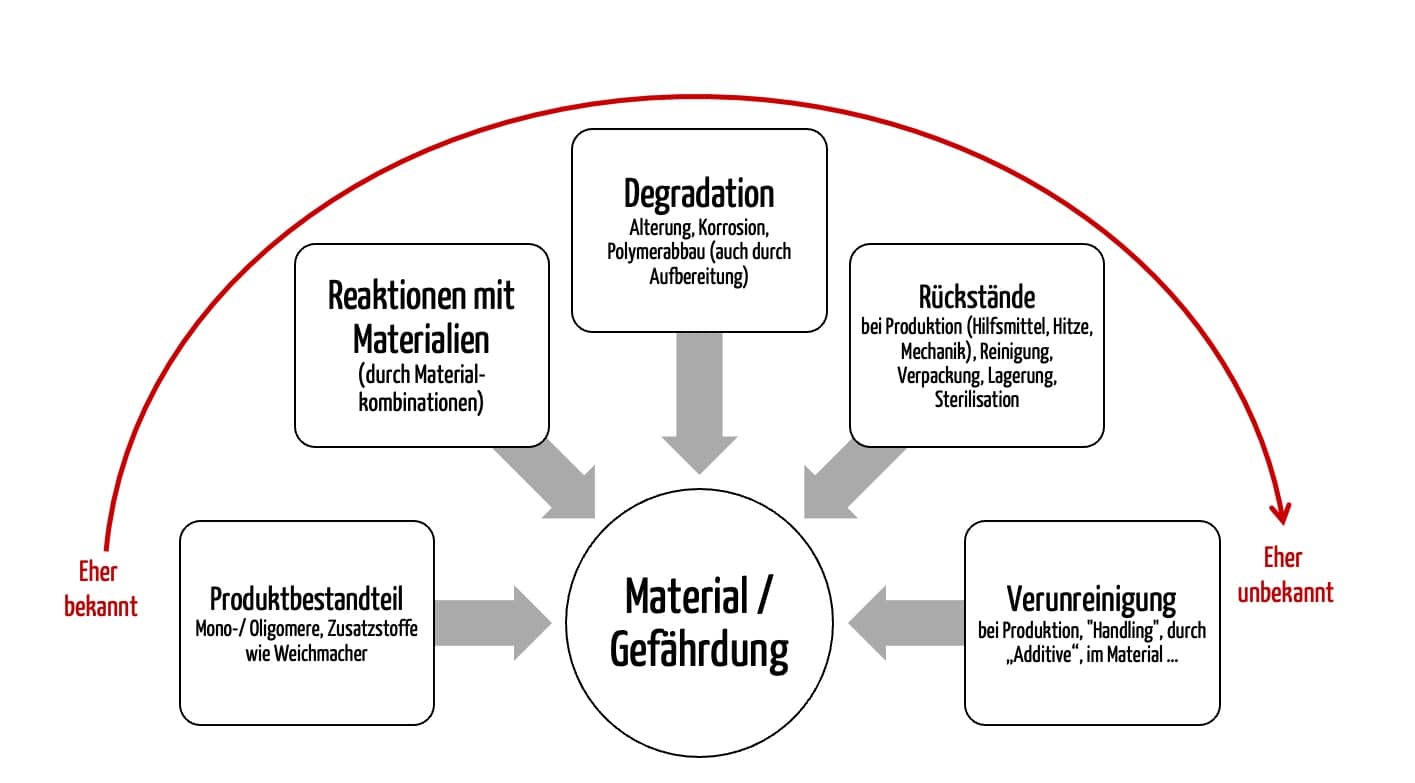
Da es neben den verwendeten Materialien weitere Faktoren gibt, die sich auf die mögliche Freisetzung von toxikologisch relevanten Stoffen auswirken und damit zu einer Gefährdung führen können, ist eine chemische Charakterisierung am finalen Medizinprodukt mit analytischen Methoden meist unerlässlich.
3. Chemische Analysen: Umsetzung nach ISO 10993-18
Risikobasierter Ansatz
Die ISO 10993-18 definiert die Anforderungen an die chemische Charakterisierung sowie konkret an die chemischen Analysen von Medizinprodukten als Teil der biologischen Bewertung. Ziel ist es, freisetzbare chemische Substanzen bzw. Stoffe zu identifizieren und quantifizieren, um potenzielle toxikologische Risiken zu bewerten.
Die ISO 10993-1 fordert dabei einen risikobasierten Ansatz, der die Art und Dauer des Patientenkontakts berücksichtigt. Die chemische Charakterisierung spielt eine zentrale Rolle, um die Teststrategie effizient anzupassen und unnötige Tierversuche zu vermeiden.
Schritt 1: Extraktion gemäß ISO 10993-18
Der erste Schritt in der chemischen Charakterisierung ist die Extraktion des Medizinprodukts. Hierbei werden chemische Substanzen unter definierten Bedingungen aus dem Material herausgelöst, um zu bewerten, welche Stoffe während der klinischen Anwendung freigesetzt werden könnten.
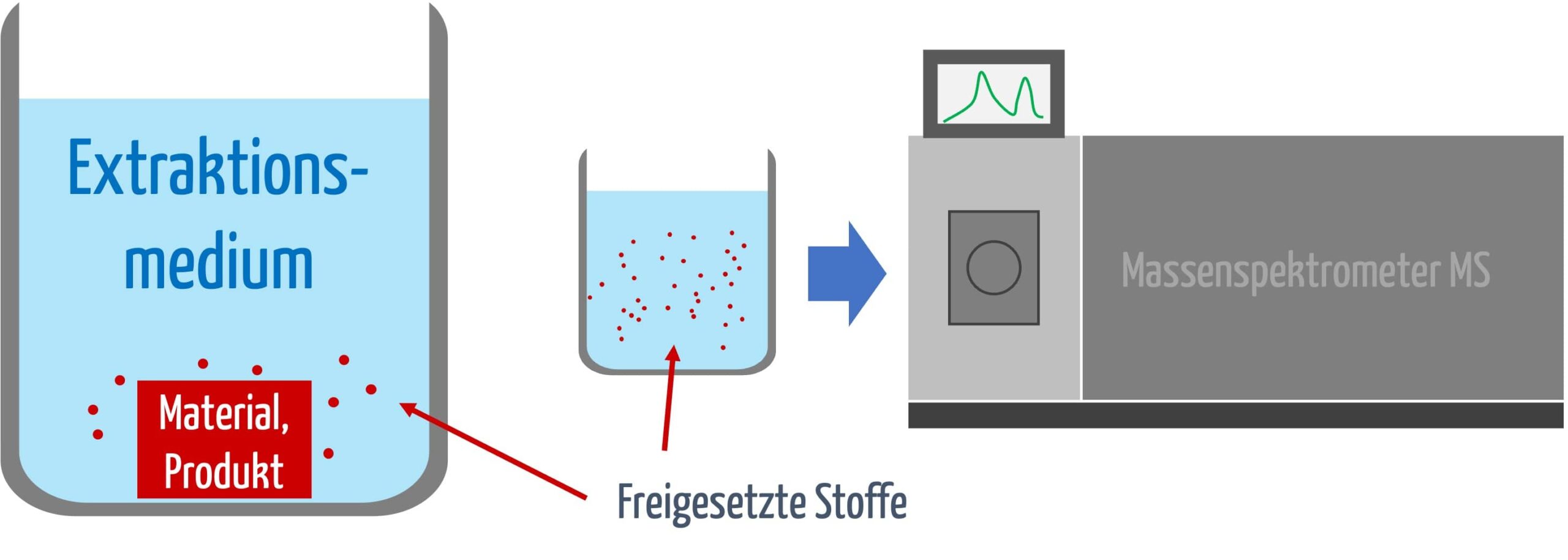
Die Art der Extraktion hängt von der Länge der Kontaktzeit ab (s. Tabelle 1).
| Kontaktkategorie | Empfohlene Extraaktionsbedingungen | Plausible Alternativen |
| Limitierter Kontakt | Simulierte Gebrauchsbedingung | Intensivierte Extraktion |
| Längerer Kontakt | Erschöpfende Extraktion | Intensivierte Extraktion |
| Langzeitkontakt | Erschöpfende Extraktion | Intensivierte Extraktion |
Neben der Kontaktzeit müssen weitere Faktoren bei der Festlegung der Extraktionsbedingungen berücksichtigt werden:
- Kumulierte Anwendung mit neuem Medizinprodukt
- Wiederverwendbare Medizinprodukte
- Materialien und Freisetzungsverhalten, z. B. nicht resorbierbares Metall
- Beständigkeit der Materialien
Wichtige Extraktionsparameter, die spezifiziert und begründet werden müssen, sind:
- Temperatur
- Dauer
- Art der Extraktionsmedien (z. B. polar, semipolar, unpolar)
- Produktoberfläche
Schritt 2: Bestimmung freigesetzter Substanzen mit analytischen Methoden
Nach der Extraktion werden chemische Analysemethoden eingesetzt, um die freigesetzten Stoffe zu identifizieren und zu quantifizieren. Die Auswahl der Methode hängt davon ab, ob die Substanzen flüchtig, halbflüchtig oder nichtflüchtig sind und ob diese anorganischer oder organischer Natur sein können.
- Gaschromatographie gekoppelt mit Massenspektrometrie (GC-MS): zur Analyse flüchtiger und halbflüchtiger organischer Verbindungen
- Hochleistungsflüssigkeitschromatographie (HPLC) oder Flüssigchromatographie mit Massenspektrometrie-Kopplung (LC-MS): zur Identifizierung nichtflüchtiger organischer Substanzen
- Induktiv gekoppeltes Plasma mit Massenspektrometrie (ICP-MS): zur Detektion anorganischer Elemente
Diese analytischen Verfahren sind entscheidend, um potenziell kritische Stoffe zu erfassen, die während der Nutzung des Medizinprodukts in den Körper gelangen könnten.
Die gewonnenen Daten fließen direkt in die toxikologische Risikobewertung nach ISO 10993-17 ein. Eine präzise chemische Charakterisierung hilft zudem, den Testaufwand für Biokompatibilitätsstudien zu reduzieren und kann dazu beitragen, Tierversuche zu vermeiden.
Die ISO 10993-18 schlägt die Prüfverfahren vor (s. Tabelle 2).
| Art der Stoffe | Beispielverfahren / Methode |
| Flüchtige organische Verbindungen | Headspace-GC-MS, GC-FID |
| Halbflüchtige organische Verbindungen | GC-MS, GC-FID |
| Nichtflüchtige organische Verbindungen | HPLC, LC-MS |
| Anorganische Elemente | ICP-MS, ICP-OES |
Die Auswahl der Methode muss auf Basis von Expositionsart und -dauer, Materialart und Freisetzungsverhalten erfolgen, um eine zuverlässige chemische Charakterisierung zu gewährleisten.
Schritt 3: Berechnung des analytischen Schwellenwerts (AET)
Der Analytical Evaluation Threshold (AET) definiert die niedrigste Konzentration einer Substanz, die zuverlässig quantifiziert werden muss, um eine toxikologische Bewertung durchzuführen.
Ziel ist es, potenziell schädliche Substanzen zu identifizieren und sicherzustellen, dass keine relevanten Risiken übersehen werden.
Bedeutung des AET
Anhand des AET können die Hersteller entscheiden, ob eine toxikologische Risikobewertung notwendig ist:
Substanzen mit einer detektierten Konzentration oberhalb des AET müssen toxikologisch bewertet werden. Hingegen gelten Substanzen in Konzentrationen unterhalb des AET als nicht sicherheitsrelevant.
Zudem bestimmt der AET die erforderliche Sensitivität der analytischen Methoden.
Berechnung des AET
Der AET wird aus mehreren Parametern berechnet:
- A: Anzahl der extrahierten Produkte
- B: Volumen des Extrakts
- C: Anzahl der Produkte in der klinischen Anwendung
- DBT: Dosisbasierter Schwellenwert, z. B. TTC (kann der Literatur entnommen werden)
- UF: Unsicherheitsfaktor

Stellen Sie sicher, dass die Empfindlichkeit der Analysen im Zusammenhang mit der geplanten Methode den Anforderungen der ISO 10993-18 entspricht. Berücksichtigen Sie den Unsicherheitsfaktor der Analyse des Labors.
Eine präzise Berechnung des AET ist entscheidend, um klinisch relevante Risiken zu identifizieren und regulatorische Anforderungen zu erfüllen.
4. Regulatorische Anforderungen
a. Europa / MDR
Die MDR fordert die biologische Sicherheit der Produkte im Anhang I. Zwar fordert die MDR nicht explizit chemische Analysen. Aber die EN ISO 10993-1 und die ISO 10993-18 sind in Europa harmonisiert und werden daher als Stand der Technik betrachtet und erwartet.
b. FDA-Guideline – Chemische Analysen
Die FDA-Guideline „Chemical Analysis for Biocompatibility Assessment of Medical Devices“ wurde 2024 in einer DRAFT-Version veröffentlicht und setzt bereits jetzt neue Maßstäbe für die Planung, Durchführung und Berichterstattung chemischer Analysen.
| Kontaktzeit | Extraktionsmethode | Lösungsmittel | NVR-Analyse erforderlich? |
| < 24 h (Limited) | Exaggerated- oder Worst-Case-Bedingung | Polar + unpolar | Nein |
| 1–30 Tage (Prolonged) | Erschöpfende oder exaggerierte Extraktion | Polar + unpolar | Ja |
| > 30 Tage (Long-Term) | Erschöpfende oder exaggerierte Extraktion | Polar, semipolar + unpolar | Ja |
Die FDA betrachtet die ISO 10993-18 als „recognized standard“. Zusätzlich erwartet sie bei chemischen Analysen:
- Testen von Triplikaten, um Produktvariabilität zu berücksichtigen
- Detaillierte Dokumentation über Probenvorbereitung und Analysemethoden
- Standardisierte Extraktionslösungsmittel und Vorgehen bei Unverträglichkeit
- Analyse nichtflüchtiger Rückstände (NVR), um die Erschöpfung der Extraktion nachzuweisen
- Umfangreiche Laborberichte (Kalibrierungsdaten, Confidence Level für Identifikation, Standards …)
Diese Anforderungen sollen sicherstellen, dass die chemischen Analysen zu Extractables reproduzierbar, validiert und regulatorisch konform durchgeführt werden.
5. Tipps zur Vermeidung von Fehlern
Fehler in der chemischen Charakterisierung nach ISO 10993-18 können zu Zulassungsverzögerungen, unnötigen Kosten und Abweichungen in Audits führen. Die folgenden Best Practices helfen, typische Fallstricke zu vermeiden und eine effiziente Biokompatibilitätsbewertung sicherzustellen.
1. Tipp: Frühzeitig planen
- Materialauswahl mit Fokus auf Biokompatibilität: Bereits in der Entwicklungsphase sollten alle chemischen Informationen zu verwendeten Materialien bei der Materialqualifikation erfasst werden (material compliance)
- Experten einbeziehen: Ein frühzeitiges Einbinden von Toxikologie-Experten kann spätere unliebsame Überraschungen vermeiden.
- Erste Screening-Analysen erwägen: So lassen sich potenzielle Risiken frühzeitig identifizieren.
2. Tipp: Methoden und Parameter systematisch festlegen
- Detaillierte chemische Informationen sammeln: Werkstoffzusammensetzung und mögliche Additive identifizieren, potenzielle Abbauprodukte und Verunreinigungen berücksichtigen
- Risikobasierten Ansatz verfolgen: Die Wahl der Analysemethoden und Parameter muss sich an Expositionsart und -dauer und den Materialeigenschaften orientieren.
- Chemische Analysen liefern Daten für die toxikologische Risikobewertung, das heißt: Eine erfolgreiche Risikobewertung ist abhängig von richtig geplanten und durchgeführten chemischen Analysen.
3. Tipp: Validierte analytische Verfahren nutzen
- Geeignetes Labor auswählen: z. B. Akkreditierung nach ISO 17025
- Labor mit einschlägiger Erfahrung zu ISO 10993-18 und FDA-Guidelines bevorzugen
- Empfohlene validierte Methoden nach ISO 10993-18 nutzen
- Sicherstellen der Empfindlichkeit genutzter Analysen mittels AET-Bestimmung
4. Tipp: Toxikologische Risikobewertung nach ISO 10993-17 durchführen
- Die freisetzbaren Stoffe müssen toxikologisch gemäß ISO 10993-17 von einem Experten bewertet werden, eine chemische Analyse allein reicht nicht aus.
- Voraussetzung für die Bewertung ist eine detaillierte Identifizierung und Quantifizierung der Stoffe.
- Es ist ein konservativer Ansatz bei der Bestimmung der tatsächlichen Exposition, der tolerierbaren Aufnahmemenge sowie der resultierenden Sicherheitsspannen (Margin of Safety) notwendig.
6. Fazit und Unterstützung
Eine gut durchdachte chemische Charakterisierung nach ISO 10993-18 spart Zeit sowie Kosten und reduziert das Risiko regulatorischer Hürden. Hersteller sollten frühzeitig eine Strategie entwickeln, um:
- Fehlende oder unzureichende chemische Daten zu vermeiden
- Teststrategien effizient zu planen und unnötige Tierversuche zu reduzieren
- Testbedingungen und Parameter smart zu wählen für eine erfolgreiche toxikologische Risikobewertung
- FDA-Anforderungen für den US-Markt jetzt schon umzusetzen
Wir begleiten Sie durch den gesamten Prozess – von der Planung bis zur Zulassung:
- Grundlagenseminar Biokompatibilität: Verständnis der regulatorischen Anforderungen
- Strategieerstellung im Biological Evaluation Plan (BEP): Risikobasierte Testplanung
- Organisation und Unterstützung bei Laborprüfungen: Sicherstellung valider Analysen
- Plausibilitätscheck / Maßnahmen bei unerwarteten Prüfergebnissen: Troubleshooting für Hersteller
- Toxikologische Risikobewertung (TRA): Bewertung gemäß ISO 10993-17
- Erstellung des Biological Evaluation Report (BER): Regulatorisch konforme Dokumentation
Kontaktieren Sie uns für eine effiziente und normgerechte Umsetzung der chemischen Charakterisierung nach ISO 10993-18!


