
In-vitro-Diagnostika (IVD) sind Medizinprodukte, mit denen Proben analysiert werden, die aus dem menschlichen Körper stammen. Typischerweise handelt es sich um Reagenzien, Kits oder Instrumente und Geräte. Auch Software kann als IVD zählen.
Bei der „Zulassung von IVD“ müssen die Hersteller viele Verordnungen, Gesetze und Normen beachten.
Der Schlagwortartikel zu den IVD enthält die Definitionen der EU und der FDA, welche Produkte als In-vitro-Diagnostikum zählen und als solches zugelassen werden müssen. Der Artikel verschafft auch einen Überblick über die regulatorischen Anforderungen an IVDs.
Ein weiterer Artikel beschreibt die 7 Schritte zum Medizinprodukt / IVD. Lesen Sie diesen Artikel zuerst.
Beachten Sie auch die Podcast-Episode. Darin berichtet Andreas Kalchschmidt-Lehmann (Molekularbiologe und ehemaliger Mitarbeiter einer Benannten Stelle) über die Hürden, an denen IVD-Hersteller regelmäßig scheitern. Und er gibt Tipps, wie diese zu meistern sind.

1. Wesentliche Schritte für die Zulassung von IVD in der EU
Die EU-Richtlinie 98/79/EG über In-vitro-Diagnostika (IVDD) wurde durch die „Verordnung (EU) 2017/746 über In-vitro-Diagnostika und zur Aufhebung der Richtlinie 98/79/EG und des Beschlusses 2010/227/EU der Kommission“ – kurz IVDR – abgelöst.
Lesen Sie mehr zur EU-Verordnung im Fachartikel zur IVDR.
Besonders schnell navigieren Sie in dieser Version der IVDR.
Falls Sie Ihr Produkt noch unter der In-vitro-Diagnostik-Richtlinie (IVDD) in den Verkehr gebracht haben, sind die IVDR-Übergangsfristen für Sie relevant.
a) Präzise Zweckbestimmung formulieren und Produkt als IVD qualifizieren
Die Zweckbestimmung eines Produkts entscheidet wesentlich über seine Qualifizierung als Medizinprodukt bzw. IVD. Sie ist auch maßgeblich für die anschließende Klassifizierung und mögliche Zulassungsstrategie. Daher sollten Hersteller in einem ersten Schritt die Zweckbestimmung ihres Produkts präzise formulieren und dabei die Anforderungen der IVDR gemäß Anhang I, Abschnitt 20.4.1 c) bzw. Anhang II, Abschnitt 1.1 c) einhalten. Auch medizinische Labore sollten für ihre Inhouse-IVD eine IVDR-konforme Zweckbestimmung schreiben.
Damit sich ein Produkt als IVD qualifiziert, muss es einen medizinischen Zweck haben. Das besagt die Definition eines Medizinprodukts gemäß MDR, Artikel 2, Nr. 1. Denn jedes IVD ist auch ein Medizinprodukt, wie die Definition eines IVDs in Artikel 2, Nr. 2 der IVDR darlegt. Kurz gesagt sind die In-vitro-Diagnostika somit eine Untergruppe der Medizinprodukte, die medizinische bzw. diagnostische Informationen aus menschlichen Proben gewinnen.
Mehr zur Qualifizierung eines IVDs lesen Sie in diesem Fachartikel.
In einem weiteren Fachartikel beschäftigen wir uns mit der Qualifizierung von Software als IVD Medical Device Software (IVD MDSW).
Die Abgrenzung zu „Research Use Only“ Produkten betrachten wir in unserem Fachartikel Laborprodukte „For Research Use Only (RUO)“ – Oft eine gefährliche Behauptung
Wenn Sie mehr zur Unterscheidung von IVD und allgemeinem Laborbedarf erfahren möchten, legen wir Ihnen unseren Fachartikel Allgemeiner Laborbedarf: Was Hersteller und Labore wissen müssen, um sich Ärger und unnötige Aufwände zu ersparen ans Herz.
b) Produkt gemäß IVDR Anhang VIII klassifizieren
Die IVDR unterscheidet vier Klassen von IVD: A, B, C und D. Die Produkte mit dem geringsten Risiko fallen in die Klasse A, die mit dem höchsten Risiko in die Klasse D.
Dieser Artikel beschreibt, wie IVD-Hersteller ihre Produkte zu klassifizieren haben.
Ein weiterer Artikel geht auf die Besonderheiten der Klassifizierung von IVD-Software ein.
c) Konformitätsbewertungsverfahren auswählen
Die IVDR beschreibt in den Anhängen IX, X und XI die Konformitätsbewertungsverfahren, welche die Hersteller abhängig von der Klasse des Produkts wählen dürfen.
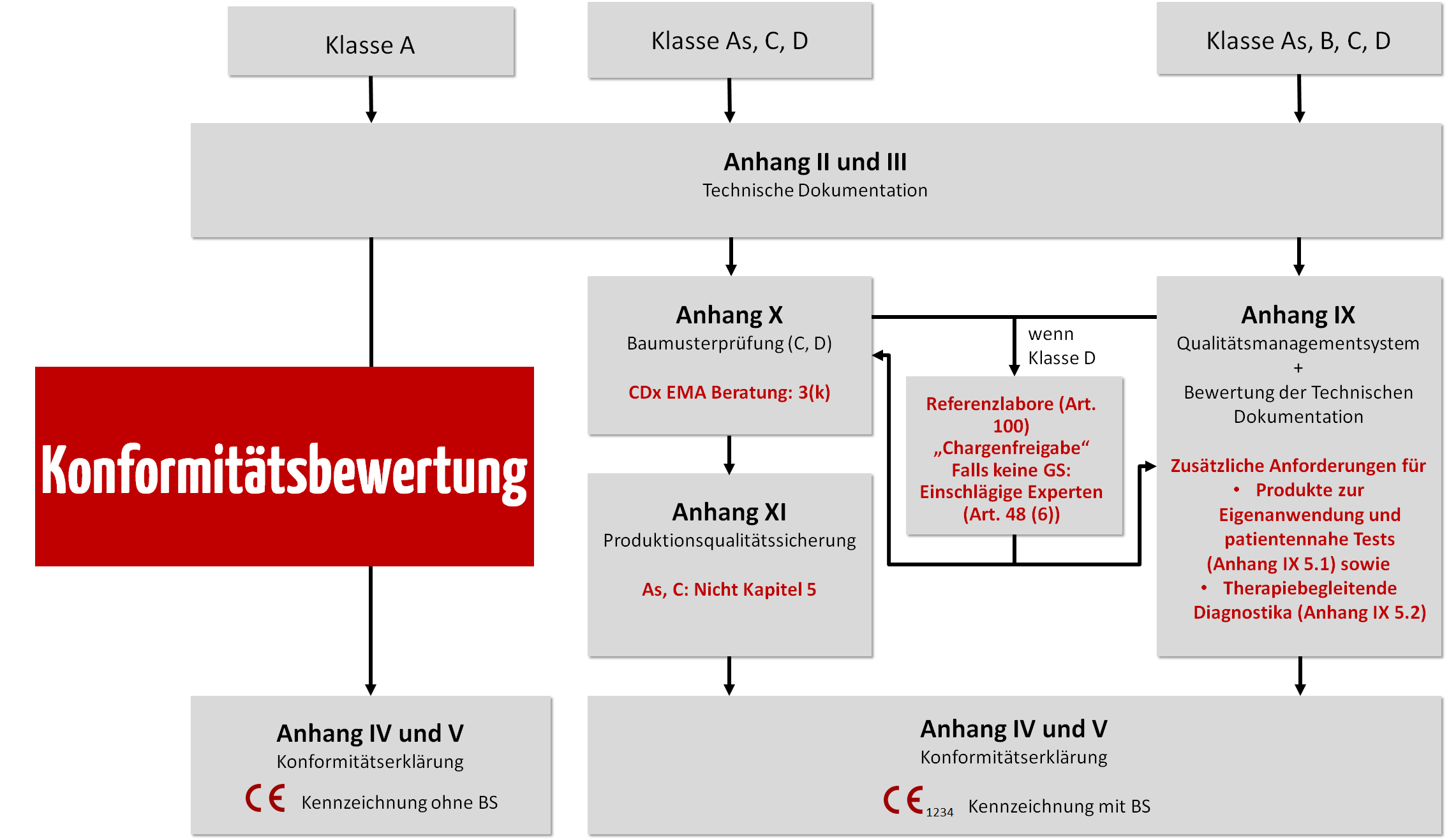
d) Anwendbare Sicherheits- und Leistungsanforderungen nachweisen und Technische Dokumentation erstellen
Unabhängig von der Klasse und dem Konformitätsbewertungsverfahren müssen alle IVD immer die grundlegenden Sicherheits- und Leistungsanforderungen gemäß Anhang I der IVDR erfüllen, und der Hersteller muss eine Technische Dokumentation konform zu den Anhängen II und III der IVDR verfassen.
Auch Inhouse-IVD müssen die grundlegenden Sicherheits- und Leistungsanforderungen nach Anhang I der IVDR erfüllen.
Hersteller weisen die Erfüllung der grundlegenden Sicherheits- und Leistungsanforderungen meist anhand harmonisierter Normen nach. Für die In-vitro-Diagnostika sind je nach Produkttyp (z. B. Reagenzien-Kit, Instrument, Software, Probengefäß) u. a. folgende Normen relevant:
- ISO 13485:2016: Medizinprodukte – Qualitätsmanagementsysteme – Anforderungen für regulatorische Zwecke
- ISO 14971: Medizinprodukte – Anwendung des Risikomanagements auf Medizinprodukte
- IEC 62366-1: Gebrauchstauglichkeit
- IEC 62304: Software
- ISO 20916: Klinische Leistungsstudien mit IVD
- EN ISO 17511: Anforderungen an die Ermittlung metrologischer Rückführbarkeit von Werten, die Kalibratoren, Richtigkeitskontrollmaterialien und Humanproben zugeordnet sind
- ISO 18113 (Teile 1 bis 5): Anforderungen an die bereitzustellenden Informationen (Symbole, Gebrauchsanweisung, Etikettierung usw.)
- ISO 23640: Haltbarkeitsprüfung von Reagenzien für in-vitro-diagnostische Untersuchungen
- EN 13641: Eliminierung oder Herabsetzung des von Reagenzien für in-vitro-diagnostische Untersuchungen ausgehenden Infektionsrisikos
- Normen mit Bezug zur Sterilität: EN 556 (Teil 1 und 2), ISO 11737 (Teil 2), ISO 13408 (Teile 1 bis 6) und ISO 14937
- IEC 61010-1, IEC 61010-2-101, IEC 61326-1, IEC 61326-2-6: Anforderungen an IVD-Geräte und Instrumente
- IEC 61310-1: Alarme
- ISO 11073: Datenübertragung von Point of Care Devices (nicht harmonisiert)
Zudem gibt es weitere produktspezifische Normen wie die ISO 15197 für Blutzuckermessgeräte oder die EN 14254, die die Anforderungen für Werkstoffe, Nennfüllmenge, Graduierung, Gestaltung, Ausführung, Sterilität, Additive und Informationen durch den Hersteller von Probengefäßen zur einmaligen Verwendung mit Ausnahme von Blutproben formuliert. Für Einmalgefäße zur venösen Blutentnahme beim Menschen gilt die EN 14820.
Neben den Normen müssen Hersteller, wenn für ihr spezifisches IVD verfügbar, Gemeinsame Spezifikationen erfüllen.
Nicht zuletzt gibt es zahlreiche Guidance-Dokumente, z. B. die CLSI-Guidances, die als Stand der Technik anerkannt und durch IVD-Hersteller z. B. bei der Leistungsbewertung berücksichtigt werden sollten.
Lesen Sie hier mehr zum Thema Verifizierung und Validierung von IVD.
In diesem Fachartikel haben wir Ihnen wichtige Informationen zur Leistungsbewertung von In-vitro-Diagnostika: In 8 Schritten zur Konformität zusammengefasst.
Wenn Sie unsicher sind, welche regulatorischen Anforderungen für Ihr IVD anwendbar sind oder wie Sie eine Nichtanwendbarkeit mit einer objektiven Rationale begründen können, dann sprechen Sie uns einfach an.
2. Anforderungen an die Zulassung von speziellen IVDs
a) Anforderungen an IVD-Geräte und -Instrumente

Neben der IVD-Verordnung (IVDR) existieren weitere, ggf. relevante europäische Richtlinien und Verordnungen, deren Anwendbarkeit für Geräte und Instrumente durch IVD-Hersteller zu prüfen ist:
- 2001/65/EU (Restriction of certain hazard substances RoHS) zur Vermeidung gefährlicher Stoffe in Elektro- und Elektronikgeräten
- 2002/96/EG bzw. das implementierende Elektro- und Elektronikgerätegesetz (ElektroG) über den Umgang mit Elektroschrott
- 1999/5/EG (Radio Equipment and Telecommunications Terminal Equipment, RTTE) zu Funkanlagen und Telekommunikation, die explizit auch bei Medizinprodukten anzuwenden ist. Die Nachfolge-Verordnung ist die 2014/53/EU.
Sonderfall: Maschinenrichtlinie bzw. Verordnung (EU) 2023/1230 über Maschinen
Einige IVD haben bewegliche Teile. Wenn es keine Schutzvorrichtungen gibt, können Anwender verletzt werden. Beispielsweise verfügen manche Laborautomaten über „Greifarme“. Vorgaben zu solchen Produkten formuliert die Maschinenrichtlinie (Richtlinie 2006/42/EG) bzw. die neue EU-Verordnung 2023/1230 über Maschinen.
Für Hersteller stellt sich die Frage, ob die IVDR oder die Maschinenrichtlinie oder die Maschinenverordnung anzuwenden ist. Gemäß Artikel 51 der Maschinenverordnung (EU) 2023/1230 wird „die Richtlinie 2006/42/EG [wird] [erst] mit Wirkung vom 14. Januar 2027 aufgehoben„. Darüber hinaus gelten die Übergangsfristen gemäß Artikel 52 der Verordnung.
Zur Anwendung von IVDR und/oder Maschinenrichtlinie führt Artikel 3 der Richtlinie aus:
„Werden die in Anhang I genannten, von einer Maschine ausgehenden Gefährdungen ganz oder teilweise von anderen Gemeinschaftsrichtlinien genauer erfasst, so gilt diese Richtlinie für diese Maschine und diese Gefährdungen nicht bzw. ab dem Beginn der Anwendung dieser anderen Richtlinien nicht mehr.“
Richtlinie 2006/42/EG, Artikel 3
Ähnlich formuliert es die Maschinenverordnung in Artikel 9:
„Werden bei einem bestimmten Produkt, das in den Anwendungsbereich dieser Verordnung fällt, die Risiken, die von den grundlegenden Sicherheits- und Gesundheitsschutzanforderungen in Anhang III erfasst werden, ganz oder teilweise durch Harmonisierungsrechtsvorschriften der Union abgedeckt, die spezifischer sind als diese Verordnung, so gilt diese Verordnung nicht für dieses Produkt, soweit diese spezifischen Rechtsvorschriften der Union diese Risiken abdecken.“
Verordnung (EU) 2023/1230, Artikel 9
Das heißt: Wenn die IVDR die Anforderungen genauer als die Maschinenrichtline bzw. -verordnung formuliert, ist für diese Anforderungen (nur) die IVDR zu berücksichtigen; für die anderen Anforderungen zusätzlich zur IVDR auch die Maschinenrichtline bzw. die Maschinenverordnung.
Erstellen Sie eine Tabelle, deren erste Spalte die „Vereinigungsmenge“ der Anforderungen der IVDR und der Maschinenrichtline bzw. -verordnung enthält. In der zweiten Spalte notieren Sie, ob Sie die Anforderungen überhaupt betreffen und falls ja, welche der beiden die spezifischeren Vorgaben enthält. In der dritten Spalte notieren Sie die Referenz auf die Umsetzung (z. B. eine Produktanforderung) bzw. deren Verifizierung.
b) Anforderungen an Betreiber von IVD bzw. an medizinische Labore mit Inhouse-IVD
Die Richtlinie der Bundesärztekammer zur Qualitätssicherung laboratoriumsmedizinischer Untersuchungen (RiliBÄK) wendet sich nicht an die Hersteller, sondern an die Betreiber von IVD. Darüber hinaus legt die ISO 15189 Anforderungen an die Qualität und Kompetenz medizinischer Laboratorien fest, die in-vitro diagnostische Untersuchungen durchführen.
Labore sollten darauf achten, dass sie nicht selbst zum Hersteller werden. Die IVDR beschreibt EU-weite regulatorische Anforderungen an „Produkte, die ausschließlich innerhalb von in der Union ansässigen Gesundheitseinrichtungen hergestellt und verwendet werden“, die sogenannten Inhouse-IVD (auch Laboratory Developed Tests, LDT genannt) (s. IVDR, Artikel 5 (5)).
Lesen Sie unseren ausführlichen Fachartikel Die EU reguliert medizinische Labore. Sind Inhouse-IVD (LDT) noch erlaubt?, um mehr über die IVDR-Anforderungen an Inhouse-IVD zu erfahren.
In unserem Webinar zur IVDR-Konformität von Inhouse-IVD erklären Ihnen unsere Experten, was es zu beachten gilt und wie Ihre nächsten Schritte aussehen sollten, damit Sie Ihre Inhouse-IVD weiterhin sorgenfrei nutzen können.
Das IVD-Team unterstützt Sie gerne bei der Einhaltung der IVDR-Vorgaben für Ihre Inhouse-IVD.
Im Seminar IVDR für medizinische Labore gibt Ihnen unser Laborexperte Ulrich Hafen einen umfangreichen Überblick über die regulatorischen Anforderungen an die Verwendung von Inhouse-IVD. Sie lernen, wie Sie Ihre bestehenden Inhouse-Tests konform betreiben und sind nach Abschluss des Seminars in der Lage, neue Inhouse-IVD anhand eines erarbeiteten Fahrplans effizient IVDR-konform zu dokumentieren.
Wenn Sie bereits wissen, dass Sie künftig zum CE-IVD-Hersteller und Inverkehrbringer werden wollen, empfehlen wir Ihnen das Seminar Technische Dokumentation nach IVDR.
c) Anforderungen an Probenahme-Sets
Gemäß der Definition eines IVDs (IVDR, Artikel 2, Nr. 2) gelten auch Probenbehältnisse als IVD.
„Probenbehältnis“ bezeichnet ein luftleeres wie auch sonstiges Produkt, das von seinem Hersteller speziell dafür gefertigt wird, aus dem menschlichen Körper stammende Proben unmittelbar nach ihrer Entnahme aufzunehmen und im Hinblick auf eine In-vitro-Untersuchung aufzubewahren
IVDR, Artikel 2, Nr. 3
Die regulatorischen Anforderungen an Probenahme-Sets erläutern wir in diesem Fachartikel.
d) Produkte zur Eigenanwendung und patientennahe Tests
Bei IVD zur Eigenanwendung (Devices for Self-Testing) und patientennahen Tests (auch Point-of-Care Tests (POCT)) stehen der besondere Nutzungskontext und die Nutzer im Vordergrund.
„Produkt zur Eigenanwendung“ bezeichnet ein Produkt, das vom Hersteller zur Anwendung durch Laien bestimmt ist, einschließlich Produkte, die für Tests verwendet werden, die Laien mittels Diensten der Informationsgesellschaft angeboten werden;
IVDR, Artikel 2, Nr. 5
„Produkt für patientennahe Tests“ bezeichnet ein Produkt, das nicht für die Eigenanwendung, wohl aber für die Anwendung außerhalb einer Laborumgebung, in der Regel in der Nähe des Patienten oder beim Patienten, durch einen Angehörigen der Gesundheitsberufe bestimmt ist
Die besonderen regulatorischen Anforderungen können Sie in dem Fachartikel Selbsttests und patientennahe Tests: Was das EU-Recht sagt nachlesen.
3. Fazit und Zusammenfassung
In Europa verläuft die Zulassung von IVD gemäß IVDR sehr vergleichbar mit der Zulassung von Medizinprodukten gemäß MDR.
Für den Erfolg der IVD-Hersteller ist eine präzise Zulassungsstrategie entscheidend, die auf einer genau formulierten Zweckbestimmung basiert. Sie sollte Antworten auf Fragen liefern wie:
- Handelt es sich um ein komplexeres System und aus welchen Produktkomponenten besteht dieses (z. B Assay, Gerät, Software)?
- Wie kann man das System aufteilen in IVD, Zubehör und Nicht-IVD, um eine möglichst einfache „Zulassung“ und eine möglichst hohe Flexibilität beim Einsatz und der Weiterentwicklung zu erreichen?
- In welche Klassen fallen die IVD und das Zubehör?
- Welche „Claims“ bzw. Leistungsanforderungen können und müssen an das Produkt gestellt oder vermieden werden?
- Wie lassen sich diese Claims bei der Leistungsbewertung am schnellsten und sichersten nachweisen?
Melden Sie sich, wenn Sie die Unterstützung der IVD-Expertinnen und Experten des Johner Instituts wünschen beim
- Formulieren der Zweckbestimmung,
- Festlegen der Zulassungsstrategie,
- Herleiten der Leistungsbewertungsstrategie oder
- Erstellen der produktspezifischen Dokumente zum Nachweis der grundlegenden Sicherheits- und Leistungsanforderungen.
Änderungshistorie
- 2024-03-07: Artikel vollständig überarbeitet
- 2023-04-22: Artikel überarbeitet, Hinweise auf die IVDD entfernt
- 2020-09-11: Anforderungen der IVDR ergänzt


Hallo,
Ich gebe Ihnen mit Ihrer Aussage nur teilweise recht, denn auch bei IVDs kann ganz klar unterschieden werden, was Verifizierung und Validierung ist. Und – Es gibt auch IVDs die sowohl im Labor als auch im PoC (Point of Care)Bereich eingesetzt werden, sodass eine Validierung in beiden Bereichen notwendig ist. Unterschiedliche Benutzergruppen, Umbegungsbedingungen und deren Einfluss auf die Präanalytik etc.
LG
P.
Ich stimme Ihnen absolut zu. Das dachte ich gesagt zu haben.
Guten Tag Herr Johner und Mitarbeiter,
auf Ihrer überaus hilfreichen Website habe ich die Liste der relevanten Normen gefunden. Bei der Suche im Beuth-Verlag bezüglich EN 13460, Bestimmung der Haltbarkeit beim Transport und Gebrauch, wird DIN EN 13460, Instandhaltung – Dokumente für die Instandhaltung; Deutsche Fassung EN 13460:2009, angezeigt. Die Suche anhand des Titels ergibt zu viele Treffer. Können Sie mir bitte weiterhelfen?
MfG
I. Riele
Sieh haben einen Zahlendreher entdeckt, liebe Frau Riele.
Es hätte EN 13640 nicht EN 13460 heißen müssen. Bitte verzeihen Sie.
Danke für Ihren Hinweis!
Beste Grüße, Christian Johner
Sehr geehrter Prof. Johner,
Sie schreiben in Ihrem überaus informativem Artikel, dass die Maschinenrichtlinie (2006/42/EG) nicht für IVD anwendbar sei.
Da wir uns die Frage nach der Gültigkeit dieser Maschinenrichtlinie für IVD auch immer wieder stets aufs Neue (und stets mit unterschiedlichem Ergebnis) stellen, möchte ich Sie nach der Grundlage / Begründung dieser Aussage fragen. Denn auch unsere benannte Stelle kommt regelmäßig mit Anforderungen der Maschinenrichtlinie bei unseren IVD Geräten daher und fordert deren Einhaltung (bzw. gleichwertiger Alternativen), und unser Bemühen die Anwendbarkeit der Maschinenrichtlinie auf IVD Geräte wegzudiskutieren sind häufig genug gescheitert. Wenn Sie eine wasserdichte und nachvollziehbare Argumentation bieten könnten, wäre das eine sehr sehr große Hilfe!
Zwischen 1998 und 2006 herrschte Klarheit über die Anwendbarkeit der Maschinenrichtlinie, denn in der alten Fassung (98/37/EG) wurden Medizinprodukte (und damit auch IVD) generell vom Anwendungsbereich dieser Richtlinie ausgenommen. Dieser Hinweis auf diese Ausnahme der Maschinenrichtlinie steht auch bis heute in der „alten“ aber immer noch gültigen IVD-Richtlinie 98/79EG, und vielleicht ist es diese historische Gegebenheit, die heute für so viel Verwirrung sorgt?!
Mit der „neuen“ Maschinenrichtlinie 2006/42/EG ist diese Ausnahme der Medizinprodukte vom Anwendungsbereich leider ersatzlos gestrichen worden.
Unsere IVD-Geräte fallen leider recht eindeutig in die Definition einer „Maschine“ laut 2006/42/EG und passen in keine Kategorie der gelisteten Ausnahmen vom Anwendungsbereich (wie Waffen oder Fortbewegungsmittel für Künstler).
Als einziger Rettungsanker verbleibt damit Artikel 3:
„Spezielle Richtlinien
Werden die in Anhang I genannten, von einer Maschine ausgehenden Gefährdungen ganz oder teilweise von anderen Gemeinschaftsrichtlinien genauer erfasst, so gilt diese Richtlinie für diese Maschine und diese Gefährdungen nicht bzw. ab dem Beginn der Anwendung dieser anderen Richtlinien nicht mehr.“
Nun äußern sich sowohl die alte IVD-Richtlinie 98/79/EG als auch die neue IVDR-Richtlinie (Pendant zur MDR) durchaus auch zu mechanischen Gefährdungen. Ob sie diese aber auch wie in Artikel 3 gefordert „genauer erfasst“ lässt sich schon anhand eines rein quantitativen Vergleichs der entsprechenden Passagen doch stark bezweifeln?!
Zudem verstehe ich den letzten Teilsatz von Artikel 3 so, dass nur diejenigen Gefährdungen vom Anwendungsbereich der Maschinenrichtlinie ausgenommen sind, denen sich die alternative Gemeinschaftsrichtlinie (hier: IVD / IVDR) genauer annimmt – damit gelten aber alle anderen Passagen / Gefährdungen der Maschinenrichtlinie dennoch?
In sehr gespannter Erwartung Ihrer Antwort verbleibe ich
mit besten Grüßen,
Martin Schroeder
Guten Tag,
erst einmal vielen Dank für die immer wieder hilfreichen und interessanten Beiträge auf ihrer Website.
Auf der Suche nach etwas ganz anderem bin ich auf eine von ihnen dargestellte Übersicht relevanter Normen für IVD gestoßen. Der Beitrag ist aus 2018.
Bei der Suche nach der Norm bin ich anstatt der ISO 13640 eher auf die ISO 23640 gestoßen, auch fehlt mir hier die ISO 14971. Sehe ich das richtig?
Nun wollte ich nachfragen, ob sie evtl. die Übersicht aktualisieren könnten. Das wäre ungemein hilfreich.
Vielen Dank.
A. Plietz
Liebe Frau Plietz, Vielen Dank, dass Sie uns darauf aufmerksam gemacht haben. Die Liste habe ich umgehend in Bezug auf die von Ihnen genannten Normen aktualisiert. Die aktuell mit der IVD-Richtlinie harmonisierten Normen finden Sie hier: https://eur-lex.europa.eu/legal-content/DE/TXT/PDF/?uri=CELEX:32020D0439&from=DE. Beste Grüße, Sebastian Grömminger
Vielen Dank Herr Grömminger.
VG
A. Plietz
Sehr geehrte Damen und Herren,
gerne möchte ich einen bereits beim BFARM registrierten IVD Corona Test in Deutschland an Krankenhäuser vertreiben.
Ich habe direkten Kontakt zum Hersteller nach China, nicht jedoch zum eingetragenen Vertreiber (dieser hat keine Exklusivrechte).
Kann ich die Tests als natürliche Person vertreiben ohne hier massive Risiken einzugehen?
Gerne möchte ich hier ein Beratungsgespräch mit Ihnen führen.
Mit freundlichen Grüßen
Daniel Prunkl
Lieber Herr Prunkel, Sie kommen damit in die Rolle des Importeurs und des Händlers. Dies hat eine gewisse Tragweite.
Ihre Anfrage habe ich an das IVD-Team weitergereicht. Wir melden uns bei Ihnen. Ihre personenbezogenen Daten habe ich aus dem Blog entfernt um den Datenschutz sicherzustellen.
Beste Grüße, Sebastian Grömminger
Sehr geehrte Damen und Herren,
Wir sind eine Firma mit Sitz in der Schweiz. Wir haben Covid-19 PCR und Antigentests, welche wir in der Schweiz vertreiben. Nun möchten wir gerne Wissen, wie wir vorgehen müssen um diese Tests als Schweizer Unternehmer in Deutschland vertreiben zu können. Können Sie uns evt. mitteilen, was alles dafür benötigt wird?
Besten Dank.
Mit freundlichen Grüßen,
M. Akkad
Lieber Herr Akkad,
vielen Dank für diese äußerst relevante Frage!
Für die Einfuhr des Tests in Deutschland muss gemäß Medizinproduktegesetz eine CE-Markierung auf dem Produkt angebracht sein und eine Konformitätserklärung gemäß Anhang III der IVD Richtlinie 98/79/EG vorliegen. Das sollte auch schon für die Schweiz zutreffen. Beachten Sie nun aber unbedingt, dass es einen EU-Repräsentanten (EC-REP) geben muss, der auf dem Produktlabel und in der Konformitätserklärung ausgewiesen sein muss, wenn der Hersteller seinen Sitz außerhalb der EU hat. Ansonsten übernimmt automatisch der Importeur (Einführer) die Haftung für das Produkt (siehe §5 MPG). Melden müssen Sie das Produkt bei DIMDI (Deutsches Institut für Medizinische Dokumentation und Information). Falls ein Swab mitgeliefert wird muss dieser allerdings konform zur MP-Richtlinie 93/42/EWG sein und braucht aufgrund des Sterilzustandes eine Benannte Stelle (CE-Zeichen mit 4-Stelliger Nummer der Benannten Stelle!).
Sehr geehrte Damen und Herren,
mit welcher Begründung findet die EU-Verordnung 1907/2006 zu Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) keine Anwendung bei IVDs?
Ich kann auf den ersten Blick den Ausschlussgrund nicht aus der REACH oder IVDR heraus lesen.
Mit freundlichen Grüßen
Steffen K
Sehr geehrter Herr K.
vielen Dank für Ihre wertvolle Nachfrage bzgl. der Anwendung der REACH auf IVD.
Konsequenz auszuschließen, dass die REACH auf IVD anwendbar ist, ist so nicht korrekt. Wir haben den betreffenden Satz im Text entfernt.
Vielmehr verweist die IVDR in Anhang I, Abschnitt 10.3 auf Artikel 59 der Verordnung (EG) Nr. 1907/2006 bzgl. der Ermittlung von in Anhang XIV der REACH gelisteten Stoffen für die die IVDR eine gezielte Risikominimierung vorsieht.
Artikel 2 der Verordnung (EG) Nr. 1907/2006 beschreibt den Anwendungsbereich bzw. schließt aus, wann die Verordnung nicht gilt.
Es lässt sich ableiten, dass die REACH z. B. zu erfüllen ist, wenn
– der Stoff oder das Gemisch die Kriterien für die Einstufung als gefährlich erfüllt.
– der Stoff persistent, bioakkumulierbar und toxisch oder sehr persistent und sehr bioakkumulierbar gemäß den Kriterien des Anhangs XIII ist.
– der Stoff in das Verzeichnis der zulassungspflichtigen Stoffe gemäß Anhang XIV aufgenommen wurde.
Auch für bestimmte ungefährliche Gemische (Art. 31 (3) REACH), die gefährliche Inhaltsstoffe über der Berücksichtigungsgrenze beinhalten, ist die REACH zu erfüllen.
Das ist ein komplexes Thema und wir hoffen, dass wir Ihnen mit unserer Antwort weiterhelfen konnten.
Herzliche Grüße,
Catharina Bertram