
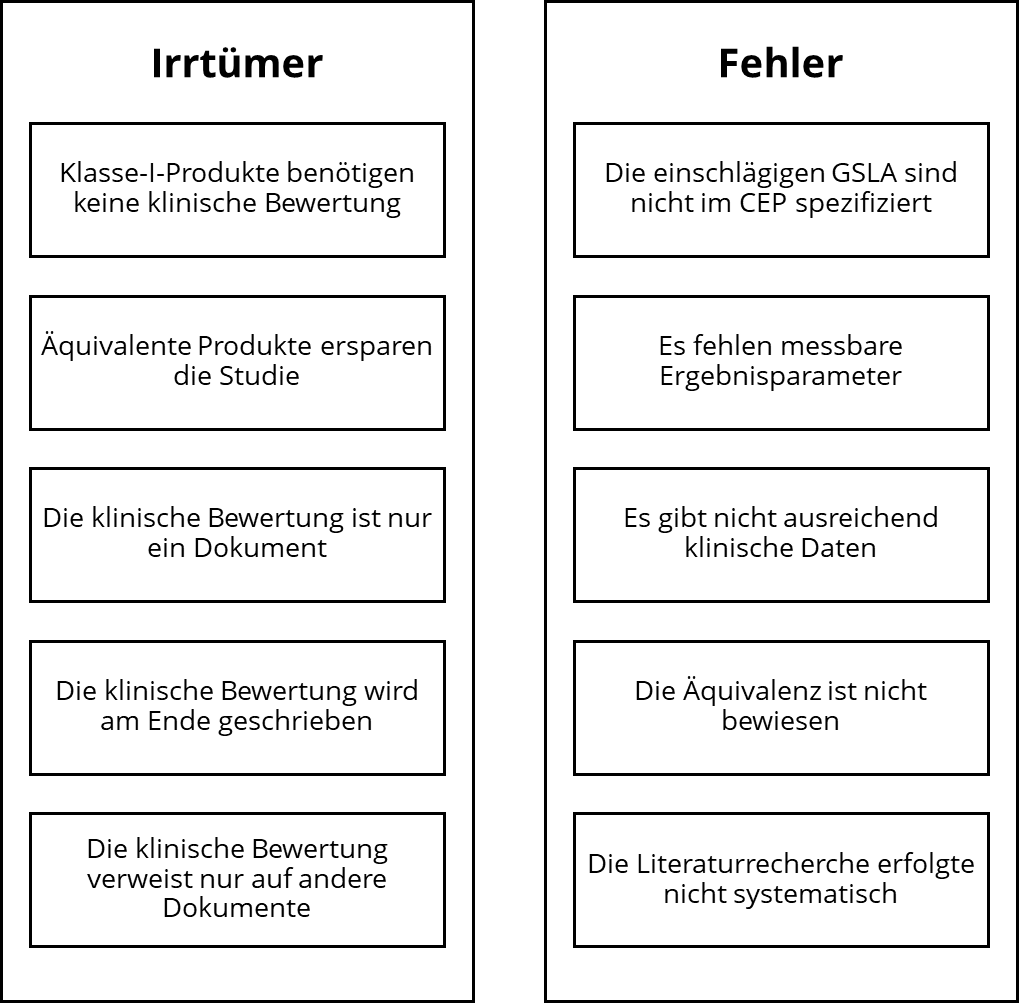
Welche fünf häufigsten Irrtümer und Fehler Medizinproduktehersteller bei der klinischen Bewertung vermeiden sollten und wie dies gelingt, zeigt dieser Artikel.
Sie stehen noch am Anfang Ihrer klinischen Bewertung?
Unser Clinical Evaluation Jumpstart Kit hilft Ihnen, die ersten Schritte innerhalb der klinischen Bewertung Ihrer Medizinprodukte zu gehen und verschafft Orientierung, was konkret von Ihnen verlangt wird.
Die fünf häufigsten Irrtümer
Irrtum 1: Ein Klasse-I-Produkt benötigt keine klinische Bewertung
Das ist falsch.
Alle Medizinprodukte müssen die grundlegenden Anforderungen der MDR (früher MDD) erfüllen. In diesem Kontext stellen beide Regularien die Forderung nach einer klinischen Bewertung.
Ein Nachweis der Einhaltung der grundlegenden Sicherheits- und Leistungsanforderungen umfasst auch eine klinische Bewertung gemäß Artikel 61.
MDR Artikel 5, Abschnitt (3)
Weder MDD noch MDR gestatten Ausnahmen, d. h. den Verzicht auf eine klinische Bewertung.
Zwar sind bei Klasse-I-Produkten keine Benannten Stellen in die Überprüfung der klinischen Bewertung involviert (auch nicht die sonstige technische Dokumentation), aber die zuständige Regierungsbehörde kann diese Bewertung jederzeit anfordern.
MDR und MDD erlauben lediglich unter ganz bestimmten Voraussetzungen, in der klinischen Bewertung auf klinische Daten zu verzichten.
Der Fachartikel zu den klinischen Daten erläutert, unter welchen Voraussetzungen Hersteller auf klinische Daten verzichten können. Nicht auf klinische Bewertungen!
Beachten Sie auch den Übersichtsartikel zu den klinischen Bewertungen.
Irrtum 2: Äquivalente Produkte ersparen die Studie (Äquivalenzroute)
Das ist fast richtig.
In der Tat erlaubt die MDR diese sogenannte „Äquivalenzroute“.
Als äquivalent gilt ein Produkt nur, wenn der Hersteller sowohl technische als auch biologische und klinische Äquivalenz nachgewiesen hat.
Die Hersteller müssen sehr detailliertes Wissen über die Äquivalenzprodukte nachweisen. Für Klasse-III-Produkte und implantierbare Produkte werden sogar Verträge unterhalb der Hersteller gefordert, die vollen Einblick in die technische Dokumentation der Äquivalenzprodukte gewähren.
Die Anforderungen an die Äquivalenzkriterien sind mittlerweile so hoch, dass sie überwiegend nur im Falle von Vorgängerprodukten aus dem eigenen Haus erfüllt werden können.
Bereits eine Änderung am Material oder an einer Beschichtung führt regelmäßig zum Verlust der biologischen Äquivalenz, eine Erweiterung der Indikationen zum Verlust der klinischen Äquivalenz etc.
Ebenfalls zutreffend ist, dass die Hersteller nicht für jedes neue Produkt automatisch eine Studie benötigen. Prüfen Sie, ob für Ihr Produkt klinische Daten für den Nachweis der grundlegenden Anforderungen an Leistung und Sicherheit überhaupt notwendig oder geeignet sind.
Lesen Sie hier mehr zum Thema Anforderungen an die Äquivalenz oder hören Sie ganz unseren Podcast zu diesem Thema.
Irrtum 3: Die klinische Bewertung ist ein Dokument
Das ist falsch.
Die klinische Bewertung ist kein Dokument, sondern ein Prozess.
„klinische Bewertung“ bezeichnet einen systematischen und geplanten Prozess zur kontinuierlichen Generierung, Sammlung, Analyse und Bewertung der klinischen Daten zu einem Produkt, mit dem Sicherheit und Leistung, einschließlich des klinischen Nutzens, des Produkts bei vom Hersteller vorgesehener Verwendung überprüft wird;
MDR Artikel 2, Absatz 44
Als Ergebnis dieses Prozesses entstehen jedoch Dokumente wie
- der klinische Bewertungsplan,
- ein Bericht zur Literaturrecherche,
- der klinische Bewertungsbericht sowie
- der Plan für den Post-market Clinical Follow-up.
Irrtum 4: Die klinische Bewertung wird erst am Ende verfasst
Das ist falsch.
Der Prozess der klinischen Bewertung kann nicht früh genug begonnen werden. Denn die klinische Bewertung ist nicht nur wichtig für die Planung, sondern auch eine wesentliche Voraussetzung beispielsweise für das Risikomanagement und die Festlegung der Risikoakzeptanzkriterien. Zudem muss die klinische Bewertung die Annahmen in der Risikomanagementakte bezüglich der klinischen Risiken bestätigen.
Richtig ist allerdings, dass der finale Bericht zur klinischen Bewertung eines der letzten Dokumente ist, das für die Zulassung eines Medizinprodukts fertiggestellt wird.
Irrtum 5: Der klinische Bewertungsbericht verweist nur auf andere Dokumente
Das ist falsch.
Der klinische Bewertungsbericht ist kein Quelldokument für Produktinformationen, sondern eine Zusammenfassung und Bewertung.
Der klinische Bewertungsbericht sollte ein eigenständiges, für sich lesbares Dokument sein.
Die Auditoren Benannter Stellen freuen sich, wenn die klinische Bewertung alle Informationen kompakt und strukturiert enthält und es ihnen erspart, diese Informationen mühsam aus vielen referenzierten Dokumenten zusammentragen zu müssen.
Es ist allerdings korrekt, dass die Dokumente der klinischen Bewertung auf andere Dokumente verweisen, z. B. auf die Risikomanagementakte, die Zweckbestimmung und die Gebrauchsanweisung.
Aber die Bewertung des Nutzen-Risikoverhältnisses, der Quantität und Qualität von Daten, der Sicherheit und Leistungsfähigkeit des Produkts ist Gegenstand der klinischen Bewertung.
Informationen wie die Zweckbestimmung müssen in allen Dokumenten den gleichen Wortlaut haben. Sie sollten immer die Referenz auf das Quelldokument angeben. Die Berichte, denen Sie die zu bewertenden Daten entnehmen, werden Sie meist zusammenfassen und ebenfalls auf die Quellen referenzieren.
Welche Informationen die benannte Stelle für den Review Ihrer klinischen Bewertung auf jeden Fall benötigt, sehen Sie im Dokument MDCG 2020-13, dem Template für den „Clinical Evaluation Report“ (CER), den der Auditor bzw. Reviewer anfertigen muss.
Bewährt hat sich die praktische MEDDEV-Checkliste des Johner Instituts.
Die fünf häufigsten Fehler
Dieser Abschnitt nennt nicht nur die fünf häufigsten Fehler, sondern gibt auch Tipps, um diese zu vermeiden.
Fehler Nr. 1: Die einschlägigen GSLA sind nicht im CEP spezifiziert
Eine häufige Kritik der Benannten Stellen lautet: „Die einschlägigen grundlegenden Sicherheits- und Leistungsanforderungen (GSLA) gemäß MDR Anhang I sind nicht im klinischen Bewertungsplan (CEP) spezifiziert.“
Die Abweichung bezieht sich auf die Anforderung der MDR in Anhang XIV, Teil A (1.a).
Erstellung und Aktualisierung eines Plans für die klinische Bewertung, der mindestens Folgendes enthält:
— Bestimmung der grundlegenden Sicherheits- und Leistungsanforderungen, die mit relevanten klinischen Daten zu untermauern sind;
MDR Anhang XIV, Teil A (1.a)
Häufig versäumen es die Hersteller, die GSLA zu bestimmen und im CEP aufzulisten.
Artikel 61 (1) der MDR fordert, dass mindestens die GSLA in den Abschnitten 1 und 8 des Anhangs I mit klinischen Daten zu untermauern sind.
Listen Sie also mindestens diese beiden GSLA im Plan der klinischen Bewertung als solche Anforderungen, die mit klinischen Daten zu untermauern sind. Prüfen Sie auch Ihre Konformitätsbewertungstabelle. Hier sollte die klinische Bewertung als Nachweisdokument mindestens unter den Leistungsanforderungen 1 und 8 auftauchen.
Die grundlegenden Sicherheits- und Leistungsanforderungen (GSLA) 1 und 8 festzulegen ist die Mindestanforderung, die Sie erfüllen müssen. Sie dürfen darüber hinaus jede GSLA hinzuziehen, die Sie als Hersteller mit klinischen Daten untermauern wollen.
Fehler Nr. 2: Es fehlen messbare Ergebnisparameter
Eine weitere Abweichung lautet: „Für den beabsichtigten klinischen Nutzen für den Patienten fehlt eine klare Definition der (messbaren) Ergebnisparameter.“
Diese Abweichung bezieht sich auf die Anforderung der MDR in Anhang XIV Teil A, 1a zum Inhalt des CEP.
Bei der Planung, der kontinuierlichen Durchführung und der Dokumentierung einer klinischen Bewertung haben Hersteller folgende Aufgaben:
a) Erstellung und Aktualisierung eines Plans für die klinische Bewertung, der mindestens Folgendes enthält:
— detaillierte Beschreibung des angestrebten klinischen Nutzens für die Patienten mit relevanten konkreten Parametern für das klinische Ergebnis;
MDR Anhang XIV, Teil A (1.a)
Der klinische Bewertungsplan sollte die konkreten Parameter für das klinische Ergebnis beschreiben. Dabei sind jene zum klinischen Nutzen festzulegen.
Ein Kühlpack soll die Schmerzen reduzieren. Der Parameter ist die Schmerzintensität mittels VAS-Score. Der Hersteller behauptet einen Rückgang um 3,5 Punkte nach 6 Wochen oder um 30 % nach 24 h.
Lesen Sie mehr dazu in unserem Fachbeitrag zu den klinischen Endpunkten.
Das MDCG 2020-6 fordert diese Parameter ebenfalls in Kapitel 6.1c und verweist zur Quantifizierung des klinischen Nutzens auf die MEDDEV 2.7/1 revision 4, Anhang A7.2, section C.
Recherchieren Sie die konkreten Parameter über die Daten zu ähnlichen Produkten, Standards oder medizinischen Leitlinien. Geben Sie hierzu ebenfalls die Schwellenwerte an, die gemäß dem Stand der Technik erreicht werden müssen.
Fehler Nr. 3: Es fehlen ausreichende klinische Daten
Häufig lautet eine Abweichung: „Es müssen ausreichende klinische Nachweise erbracht werden, um die Leistungsfähigkeit und Sicherheit jeder Indikation zu belegen.“
In diesen Fällen ist der Reviewer nicht überzeugt, dass Sie über ausreichende klinische Daten verfügen, um Ihre Behauptungen zum Medizinprodukt zu beweisen. Gemeint sind unzureichende Nachweise der GSLA 1 und 8 (Sicherheit, klinischen Leistung, klinischer Nutzen) auf Grundlage klinischer Daten.
Artikel 61 (1) im zweiten Abschnitt der MDR fordert von Herstellern die Spezifizierung des Umfangs des klinischen Nachweises:
Der Hersteller spezifiziert und begründet den Umfang des klinischen Nachweises, der erforderlich ist, um die Erfüllung der einschlägigen grundlegenden Sicherheits- und Leistungsanforderungen zu belegen. Der Umfang des klinischen Nachweises muss den Merkmalen des Produkts und seiner Zweckbestimmung angemessen sein.
MDR Artikel 61 (1), zweiter Abschnitt
Das heißt, dass Sie als Hersteller festlegen müssen, welche klinischen Evidenz ausreichend ist, um die Sicherheit, die Leistung und den klinischen Nutzen Ihres Produktes nachzuweisen. Selbstverständlich ist diese Festlegung zu begründen.
Nutzen Sie als Hilfestellung zur Festlegung des klinischen Nachweises das MDCG 2020-6. Anhang III beinhaltet eine Tabelle, die Ihnen einen Überblick gibt, welche Daten welche Aussagekraft haben können.
Beachten Sie dazu den Beitrag MDCG 2020-6: Anforderungen an klinische Daten für Legacy Devices.
Zusätzlich müssen Sie den Stand der Technik zu Ihrem Produkt berücksichtigen.
- Zählt Ihr Produkt zu einer gut etablierten Technologie?
Dann ist ein Datensatz aus PMS-Daten und Anwenderbefragungen möglicherweise ausreichend. - Ist Ihr Produkt innovativ und das Risiko-Nutzenverhältnis ist bisher nicht ausführlich beschrieben?
Dann kann eine prospektive klinischen Prüfung nötig werden. Prüfen Sie in jedem Fall, welche klinischen Endpunkte Sie festgelegt haben und ob Sie diese mit entsprechenden Datensätzen nachweisen oder nicht.
Fehler Nr. 4: Die Äquivalenz ist nicht bewiesen
Ein weiterer „Klassiker“ bei den Abweichungen ist diese: „Die Äquivalenz zwischen den Produkten ist nicht nachvollziehbar. Die Dokumentation zum Äquivalenznachweis ist nicht ausreichend.“
Eine solche Abweichung ist häufig auf eine unzureichende Bewertung der Äquivalenz zwischen zwei Produkten zurückzuführen. Die MDR fordert in Anhang XIV, Teil A (3), dass äquivalente Produkte (gleichartige Produkte) in ihren klinischen und biologischen Merkmalen nahezu gleich und technische Charakteristika vergleichbar sein müssen.
Eine nicht ausreichende Betrachtung der klinischen, technischen und biologischen Merkmale kann folgende Gründe haben:
- Sie haben Anhang I der MDCG 2020-5 nicht berücksichtigt.
- Sie haben keine wissenschaftlich fundierten Vergleiche durchgeführt.
- Sie verfügen nicht über genügend Daten zum äquivalenten Produkt, die einen Vergleich ermöglichen.
Punkt Nr. 3 ist der häufigste Grund, weshalb eine Äquivalenzbetrachtung scheitert. In den meisten Fällen fehlen:
- Daten zu bestimmten Leistungsspezifikationen
- Informationen zur Zweckbestimmung
- Daten zu den Materialien und den herauslösbaren Stoffen der äquivalenten Produkte
Unsere Tipps für einen erfolgreichen Äquivalenzvergleich:
- Sammeln Sie zunächst sämtliche Daten zum Äquivalenzprodukt.
- Führen Sie den Äquivalenzvergleich gemäß Anhang I der MDCG 2020-5 durch.
- Identifizieren Sie evtl. vorliegende Datenlücken.
- Beauftragen Sie ggf. Tests, um die Datenlücken zu füllen (Tests mit dem Äquivalenzprodukt zu bspw. dem Abgabeverhalten herauslösbarer Stoffe nach ISO 10993-18).
- Füllen Sie die Datenlücken.
- Schließen Sie den Äquivalenzvergleich ab.
Sollten die oben genannten Maßnahmen nicht umsetzbar sein oder die Äquivalenz widerlegen, können Sie die klinischen Daten zu den Produkten in den Stand der Technik überführen. Das Produkt wird dann zu einem „ähnlichen Produkt“.
In dem Fall, dass Ihnen die Äquivalenz zu einem Produkt wegbricht, können Sie die Daten zum ähnlichen Produkt nicht mehr für den Nachweis von Sicherheit und Leistung Ihres eigenen Produkts verwenden. Im Kapitel zum Stand der Technik helfen Ihnen die Daten zu ähnlichen Produkten jedoch dabei,
- die Angemessenheit des Risikomanagements zu bestätigen,
- alternative Behandlungsmethoden und Technologien zu beschreiben,
- Ihre PMCF-Aktivitäten zu planen,
- klinische Ergebnisparameter zu identifizieren sowie
- Akzeptanzkriterien und Schwellenwerte festzulegen.
Fehler Nr. 5: Die Literaturrecherche erfolgte nicht ausreichend systematisch
Viele Hersteller erhalten diese Abweichung: „Es ist nicht nachvollziehbar, ob die Literaturrecherche innerhalb der klinischen Bewertung einem methodisch fundierten Verfahren folgt.“
In diesem Fall ist die Dokumentation zur Literaturrecherche möglicherweise nicht vollständig.
Beachten Sie den Fachbeitrag Literatursuche bei der klinischen Bewertung: 6 Tipps, die Ihnen Zeit und Ärger ersparen
Die Literatursuche innerhalb der klinischen Bewertung benötigen Sie für zwei regulatorische Kontexte: die Produktsuche selbst und die Suche zum Stand der Technik. Beide Suchen sind systematisch durchzuführen und zu dokumentieren.
Die MDR fordert:
„Eine klinische Bewertung erfolgt nach einem genau definierten und methodisch fundierten Verfahren, das sich auf folgende Grundlagen stützt: a) eine kritische Bewertung der einschlägigen derzeit verfügbaren wissenschaftlichen Fachliteratur über Sicherheit, Leistung, Auslegungsmerkmale und Zweckbestimmung des Produkts […]“
MDR Artikel 61 (3)
Die MEDDEV 2.7/1 revision 4, A5 und das MDCG 2020-13, section D nennen als fundierte Verfahren der Literaturrecherche und Bewertung:
- PICO (patient characteristics, type of intervention, control, and outcome queries)
- Cochrane Handbook for Systematic Reviews of Interventions
- PRISMA (The Preferred Reporting Items for Systematic Reviews and Meta-Analyses) Statement
- MOOSE Proposal (Meta-analysis Of Observational Studies in Epidemiology)
Nutzen Sie innerhalb der klinischen Bewertung die hier genannten Methoden, um Ihre Literaturrecherche aufzubauen. Dokumentieren Sie die angewendeten spezifischen Methoden in Ihrem Literatursuchbericht. Damit sorgen Sie für eine nachvollziehbare Vorgehensweise.
Fazit & Zusammenfassung
Die klinische Bewertung ist einer der anspruchsvollsten Prozesse. Daher überrascht es nicht, dass Benannte Stellen besonders viele Fehler finden.
Kennt man die typischen Irrtümer und Fehler kennen, so lassen sich diese vermeiden.
Haben Sie Fragen zur konkreten Behebung einzelner Abweichungen? Schreiben Sie uns eine E-Mail oder buchen Sie einen Beratungstermin.
Die Clinical Experts der Johner Instituts können Sie in jedem Fall dabei unterstützen, mit einer professionellen klinischen Bewertung Ihre Audits, Reviews und Zulassungsverfahren schnell und erfolgreich abzuschließen.


