
Für die klinische Bewertung von Software gelten die gleichen gesetzlichen Anforderungen wie für die klinische Bewertung aller Medizinprodukte. Das heißt, als Hersteller von Medical Device Software (MDSW) müssen Sie genau wie alle anderen Hersteller eine klinische Bewertung für Ihr Produkt erstellen. Für Software, die ein In-vitro-Diagnostikum (IVD) darstellt, muss eine Leistungsbewertung durchgeführt werden.
Das wiederum erfordert klinische Daten zu dem Produkt, vielleicht aus einer klinischen Prüfung bzw. einer klinischen Leistungsstudie, oder zu einem nachgewiesenen Äquivalenzprodukt. Aber für Software ergibt dieser Ansatz oft wenig Sinn.
Doch das MDCG-Dokument MDCG 2020-1: Guidance on Clinical Evaluation (MDR)/Performance Evaluation (IVDR) of Medical Device Software zeigt einen möglichen alternativen Weg für Software-Produkte auf. Die Benannten Stellen haben nun erste klinische Bewertungen von Software akzeptiert, die von uns auf Basis dieses Dokuments erstellt wurden.
Sie stehen noch am Anfang Ihrer klinischen Bewertung?
Unser Clinical Evaluation Jumpstart Kit hilft Ihnen, die ersten Schritte innerhalb der klinischen Bewertung Ihrer Medizinprodukte zu gehen und verschafft Orientierung, was konkret von Ihnen verlangt wird.
1. Kurze Wiederholung: Was ist eine klinische Bewertung bzw. eine Leistungsbewertung?
„klinische Bewertung“ bezeichnet einen systematischen und geplanten Prozess zur kontinuierlichen Generierung, Sammlung, Analyse und Bewertung der klinischen Daten zu einem Produkt, mit dem Sicherheit und Leistung, einschließlich des klinischen Nutzens, des Produkts bei vom Hersteller vorgesehener Verwendung überprüft wird
Quelle: MDR Artikel 2
„Leistungsbewertung“ bezeichnet eine Beurteilung und Analyse von Daten zur Feststellung oder Überprüfung der wissenschaftlichen Validität, der Analyseleistung und gegebenenfalls der klinischen Leistung eines Produkts
Quelle: IVDR Artikel 2
Damit werden die Ziele zumindest indirekt genannt: Es geht um die Prüfung, ob das Medizinprodukt
- den Nutzen stiftet, den der Hersteller verspricht,
- sicher ist und keine Risiken birgt, die in der Risikoanalyse nicht bereits identifiziert und als akzeptabel bewertet wurden, und
- die Leistung erbringt, die der Hersteller verspricht.
Lesen Sie hier weitere Artikel über die klinische Bewertung sowie über die Leistungsbewertung von IVD.
2. Klinische Bewertung von Software: Die Besonderheiten
a) Der wesentliche Unterschied: Die Schnittstellen
Während andere Medizingeräte viele Formen an „Outputs“ (= Schnittstellen nach außen) haben, wie elektrische Energie (z.B. HF-Chirurgie-Gerät), elektromagnetische Energie (z.B. CT) oder mechanische Energie (z.B. Ultraschallgerät), ist der „Output“ von Stand-alone-Software auf Informationen beschränkt.
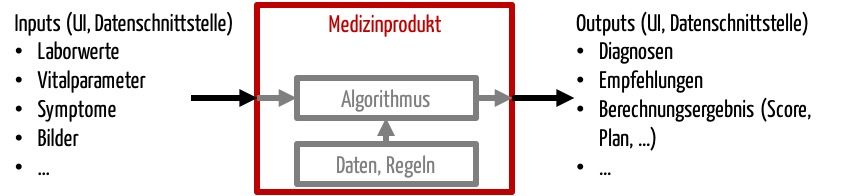
Software unterscheidet sich von anderen Medizinprodukten also durch die Schnittstellen:
- Benutzer-Produkt-Schnittstelle, oft grafische Benutzungsschnittstelle (GUI)
- Produkt-Produkt-Schnittstelle, insbesondere Datenschnittstellen
Daher muss die klinische Bewertung bei Software prüfen, ob die Informationen dem Anwender einen Nutzen liefern, beispielsweise bei Diagnose oder Therapieauswahl, oder ob sie nützliche Informationen an andere Produkte weitergibt.
Dieses Konzept kommt IVD-Herstellern sicherlich bekannt vor. So liegt der Nutzen eines IVDs stets in der Bereitstellung angemessener medizinischer Informationen (s. IVDR, Erwägungsgrund 64). Der klinische Nachweis wird für die in-vitro diagnostische Software durch die Leistungsbewertung erbracht.
Den Fall, dass die Software zur Ansteuerung von Medizinprodukten dient, betrachten wir hier nicht. Denn die Software wäre in den meisten Fällen ein Zubehör zu dem jeweiligen Produkt, zu dem bereits eine klinische Bewertung existieren sollte.
b) Besonderer Nutzen von Stand-alone-Software
Stand-alone-Software dient v.a. einem der folgenden Zwecke:
- Diagnose oder Bereitstellung diagnoserelevanter Informationen (Medizinprodukte/IVD)
- Auswahl geeigneter Therapien, z.B. Auswahl eines geeigneten Medikaments oder Entscheidung, ob Chemotherapie oder Bestrahlung (Medizinprodukte/IVD)
- Durchführen der Therapie selbst, z.B. Berechnung des Einstichwinkels eines chirurgischen Instruments, Bestrahlungsplanung, Dosisberechnung (nur Medizinprodukte)
Das Guidance-Dokument MDCG 2019-11 „Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 – MDR and Regulation (EU) 2017/746 – IVDR“ liefert Vorgaben zur Unterscheidung von Software als Medizinprodukt bzw. IVD.
c) Klinische Bewertung bzw. Leistungsbewertung anhand Verifizierung und Validierung
Die klinischen Bewerter und Leistungsbewerter stellen sich bei Softwareprodukten häufig die Frage, welche Aspekte sie überhaupt bewerten müssen. Meistens ist es sinnvoll, sich auf die Algorithmen zu konzentrieren, z.B.:
- Lässt ein Bildbearbeitungsalgorithmus Tumore mit ausreichender Wahrscheinlichkeit erkennen?
- Hilft der Algorithmus eines Arzneimitteltherapie-Sicherheitssystems, Kontraindikationen und Wechselwirkungen zu erkennen und Fehlmedikationen zu vermeiden?
Manchmal sind diese Algorithmen banal. Dann stellt sich die Frage, was eine klinische Bewertung bzw. Leistungsbewertung von Software an Erkenntnissen bringen soll. In manchen Fällen kann es ausreichen, anhand von Verifizierungsergebnissen zu argumentieren.
Falls die Algorithmen hingegen komplexer sind, bedarf es des Beweises, dass sie den versprochen Nutzen bringen, d.h. klinisch valide sind. Genau diesen Ansatz verfolgt das MDCG-Dokument MDCG 2020-1.
3. Mit drei Beweisen zur klinischen Bewertung von Software laut MDCG 2020-1
Laut MDCG muss der Hersteller drei Beweise erbringen:
- Wissenschaftliche Validität („scientific validity“)
- Technische/analytische Leistungsfähigkeit („technical performance/analytical performance“)
- Klinische Leistungsfähigkeit („clinical performance“)
Bei der Beweisführung sollte der Hersteller dabei fünf Phasen durchlaufen:
- Planung
- Datensammlung
- Bewertung der Daten
- Analyse der Daten
- Dokumentation
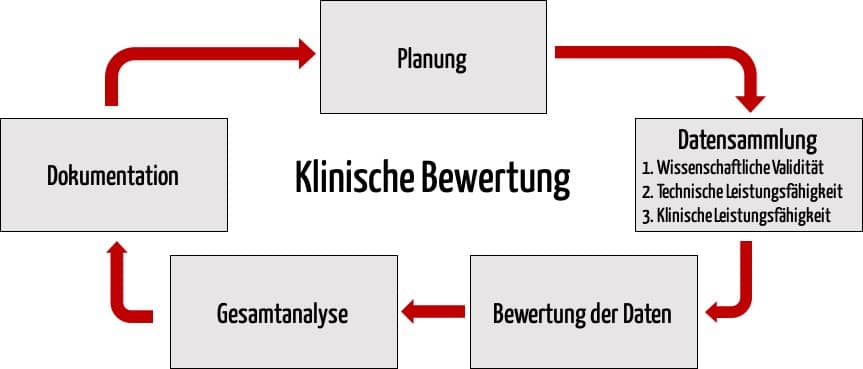
Diese drei Beweistypen werden im Folgenden vorgestellt.
a) Beweis 1: Wissenschaftliche Validität
Quelle MDCG 2020-1
„…is understood as the extent to which, the MDSW’s output (e.g. concept, conclusion, calculations) based on the inputs and algorithms selected, is associated with the targeted physiological state or clinical condition. This association should be well founded or clinically accepted“
Quelle: MDCG 2020-1
Die wissenschaftliche Validität ist also der Evidenzbeweis, dass die „Outputs“ des Produkts (hier der Software) mit einem klinischen oder physiologischen Zustand verknüpft sind.
Ein Beispiel stellt die Berechnung eines Scores dar, der tatsächlich ein Maß für ein bestimmtes Risiko darstellen muss.
Zum Nachweis der wissenschaftlichen Evidenz sollen die Hersteller folgende Quellen nutzen:
- Wissenschaftlicher Fachliteratur („peer reviewed articles“, Konferenzartikel)
- Empfehlungen der Behörden
- Wissenschaftliche Datenbanken, auch mit Ergebnissen klinischer Studien
- Expertenmeinungen, z.B. in Büchern und Leitfäden
b) Beweis 2: Technische Leistungsfähigkeit
„… is the demonstration of the MDSW’s ability to accurately, reliably and precisely generate the intended output, from the input data.“
Quelle: MDCG 2020-1
Die Verifizierung der technischen Leistung ist somit die Demonstration der Fähigkeit des Software-Produkts, aus den Eingabedaten genau, zuverlässig und präzise die beabsichtigte Ausgabe zu erzeugen.
Die technische Leistungsfähigkeit beweisen Hersteller im Rahmen der Verifizierung und Validierung (V&V). Dabei sollen sie ggf. zurückgreifen auf:
- 1. Verifizierungsaktivitäten, z.B.:
- Unit-Tests
- Integrationstests
- Systemtests
- 2. Validierungsaktivitäten, z.B. anhand von:
- Kuratierten Datenbanken oder Registern
- Referenzdatenbanken
- Bereits gesammelten Patientendaten
Üblicherweise referenzieren Medizinprodukte-Hersteller bei der klinischen Bewertung von Software diese V&V-Ergebnisse.
IVD-Hersteller bewerten die technische Leistungsfähigkeit der Software abschließend während der analytischen Leistungsbewertung des IVDs und berücksichtigen vorliegende V&V-Ergebnisse.
c) Beweis 3: Klinische Leistungsfähigkeit
„…is the demonstration of a MDSW’s ability to yield clinically relevant output in accordance with the intended purpose“
Quelle: MDCG 2020-1
Der Hersteller muss bei der klinischen Leistungsfähigkeit eine klinische Relevanz in Bezug auf die Zweckbestimmung des Produkts nachweisen. Die klinische Relevanz einer Medical-Device-Software zeigt sich als positive Auswirkung auf
- die Gesundheit eines Individuums, ausgedrückt in Form von messbaren, patientenrelevanten klinischen Endpunkten einschließlich Endpunkt(en) im Zusammenhang mit der Diagnose, Risikovorhersage, Vorhersage der Behandlungsreaktion(en), oder
- Aspekte, die mit seiner Funktion zusammenhängen, z.B. die der Früherkennung, Überwachung, Diagnose oder Hilfe zur Diagnose von Patienten, oder
- das Patientenmanagement oder zur öffentlichen Gesundheit.
Diese Daten können ebenso aus vom Hersteller durchgeführten klinischen Studien stammen wie auch aus anderen Quellen. Die Hersteller werden ermutigt, auch auf bestehende Daten von alternativen Verfahren und Produkten zurückzugreifen.
Sie sollten z.B. die folgenden Informationen heranziehen, um die klinische Leistungsfähigkeit zu beurteilen:
- Klinische/diagnostische Sensitivität und/oder klinische/diagnostische Spezifität
- Positiver/negativer prädiktiver Wert
- Number needed to treat (Anzahl der Patienten, die aufgrund einer Erkrankung oder präventiv behandelt werden müssen, um ein zusätzliches Ereignis wie (Folge-) Krankheit oder Tod zu vermeiden)
- Number needed to harm (Anzahl der Patienten, die diagnostiziert/behandelt werden müssen, um eine unerwünschte Wirkung auf einen Patienten zu haben)
- Benutzerfreundlichkeit/Benutzeroberfläche
- Konfidenzintervall(e)
d) Zusammenfassung der Schritte für die klinische Bewertung von Software
Das folgende Ablaufschema zeigt noch einmal die wesentlichen Schritte der Erstellung der klinischen Bewertung:

4. Klinische Bewertung von Software nach der Inverkehrbringung (Post-Market)
Die Hersteller müssen nach der Inverkehrbringung die Sicherheit, die Wirksamkeit und die Leistung des MDSW aktiv und kontinuierlich überwachen.
Wie bei allen Herstellern können die Informationen u.a. Beschwerde, direktes Endbenutzer-Feedback oder neu veröffentlichte Forschungen/Richtlinien umfassen. Ganz besonders relevant für MDSW ist aber die Erfassung von REAL-WORLD PERFORMANCE DATA, da diese mit Software relativ leicht zu gewinnen sind.
Dies ermöglicht sehr schnell und effektiv
- die rechtzeitige Erkennung und Korrektur von Fehlfunktionen,
- die Erkennung von systematischem Missbrauch,
- ein Verständnis der Benutzerinteraktionen und
- die laufende Überwachung der vorgesehenen klinischen Leistung (z.B. Sensitivität, Spezifität).
Sie finden hier Informationen zur Post-Market Surveillance und zum Post-Market Surveillance Plan.
5. Beurteilung des Evidenz-Niveaus und der Daten
Die Hersteller müssen in erster Linie selbst entscheiden, ob für jeden Schritt das Niveau der Daten ausreichend ist und dies ggf. auch begründen. Hierbei geht es sowohl um die Menge als auch um die Qualität der Daten. Diese Bewertung kann sich an den folgenden Fragen orientieren:
a) Ausreichende Menge
- Unterstützen die Daten den Verwendungszweck, die Indikationen, die Zielgruppen, die klinischen Behauptungen und die Kontraindikationen?
- Sind die klinischen Risiken und die analytische Leistung bzw. klinische Leistung untersucht worden?
- Wurden relevante MDSW-Merkmale wie die Dateneingabe und -ausgabe, die angewandten Algorithmen oder die Art der Zusammenschaltung bei der Generierung der Daten zur Unterstützung der Leistung des Produkts berücksichtigt?
- Wie hoch ist der Innovationsgrad bzw. die Geschichte auf dem Markt (Wie groß ist der Bestand an wissenschaftlichen Erkenntnissen)?
b) Ausreichende Qualität
- Waren die Art und das Design der Studie bzw. des Tests geeignet, die Endpunkte zu erreichen?
- War der Datensatz angemessen und aktuell (Stand der Technik)?
- War der statistische Ansatz angemessen, um zu einer gültigen Schlussfolgerung zu gelangen?
- Wurden alle ethischen, rechtlichen und regulatorischen Erwägungen bzw. Anforderungen berücksichtigt?
- Besteht ein Interessenkonflikt?
c) Nachweis der Konformität ohne klinische Daten
In einigen Fällen, insbesondere bei Medical-Device-Software der niedrigeren Risikoklassen I und IIa, sind klinische Daten zum Nachweis der Konformität oft nicht angemessen. Sowohl die MDR (Artikel 61, Abschnitt 10) also auch das MDCG-Dokument 2020-1 erlauben eine Ausnahme.
Der Nachweis der Übereinstimmung mit grundlegenden Sicherheits- und Leistungsanforderungen kann allein auf der Grundlage der Ergebnisse nichtklinischer Testmethoden einschließlich Leistungsbewertung, technischer Prüfung („bench testing“) und vorklinischer Bewertung erfolgen. Dies muss aber auf jeden Fall begründet werden. Dabei müssen die folgenden Punkte berücksichtigt werden:
- Risikomanagements des Herstellers
- Berücksichtigung der besonderen Merkmale des Zusammenspiels zwischen dem Produkt und dem menschlichen Körper
- Bezweckte klinischen Leistung und Angaben des Herstellers zur Leistung
ACHTUNG: Die Hersteller müssen trotzdem eine klinische Bewertung erstellen, die zumindest die Begründung für den Ausnahmeweg, den Nachweis der nichtklinischen Testmethoden sowie die Darlegung des State of the Art enthält.
Auch für In-vitro-Diagnostika gilt, dass, wenn bestimmte Leistungsanforderungen nicht anwendbar sind, diese begründet ausgeschlossen werden können. Die IVD-Hersteller beschreiben ihre beabsichtigte Strategie zur Leistungsbewertung im produktspezifischen Leistungsbewertungsplan. Dort legen sie auch den Stand der Technik dar. Im Leistungsbewertungsbericht werden die Ergebnisse zum klinischen Nachweis und zum Nutzen-Risiko-Verhältnis abschließend zusammengefasst und bewertet.
6. FDA & IMDRF zur Clinical Evaluation von Software as a Medical Device (SaMD)
Gemeinsam mit dem IMDRF hat die FDA bereits ein sehr ähnliches Guidance-Dokument „Software as a Medical Device: Clinical Evaluation“ publiziert. Es fordert, dass die Hersteller mit der klinischen Bewertung von Medizinprodukte-Software bzw. mit der Leistungsbewertung von IVD-Software die klinische Validität des Produkts überprüfen. Damit weisen sie nach, dass die beabsichtigte Zweckbestimmung erfüllt ist und keine inakzeptablen Risiken bestehen. Einige der Definitionen weichen von denen der MDCG ab. Das führt unter Umständen zu einem hohen Mehraufwand, falls beide Länder bedient werden müssen.
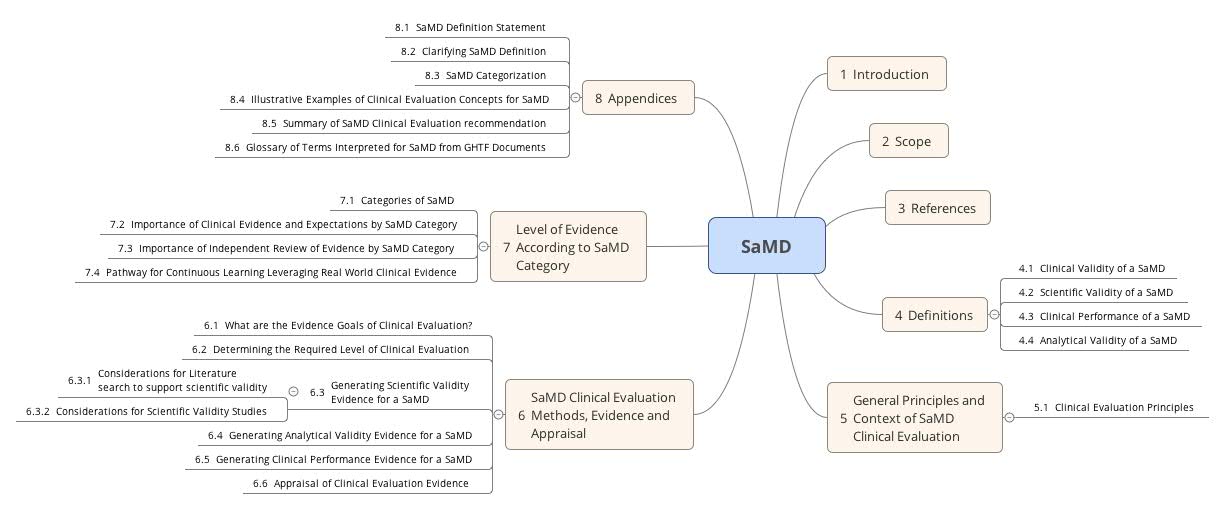
Die FDA gibt Hinweise, welche Schritte in Abhängigkeit von gewissen Parametern (Risikoklasse, Innovationsgrad etc.) gemacht werden müssen.
a) Klasse für die Software bestimmen
Die FDA verwendet für die klinische Bewertung von Software eine Klassifizierung aus anderen IMDRF Dokumenten:
| Significance of information provided by SaMD to healthcare decision | |||
|---|---|---|---|
| State of healthcare situation or condition | Treat or diagnose | Drive clinical management | Inform clinical management |
| Critical | IV.i | III.i | II.i |
| Serious | III.ii | II.ii | I.ii |
| Non-serious | II.iii | I.iii | I.i |
Als Beispiele für die Anwendungsgebiete nennt die FDA:
- „Treat or diagnose“
- Treat
- Provide therapy to a human body using other means
- Diagnose
- Detect
- Screen
- Prevent
- Mitigate
- Lead to an immediate or near term action
- „Drive clinical management“
- Aid in treatment
- Provide enhanced support to safe and effective use of medicinal products
- Aid in diagnosis
- Help predict risk of a disease or condition
- Aid to making a definitive diagnosis
- Triage early signs of a disease or condition
- Identify early signs of a disease or condition.
- „Inform clinical management“
- Inform of options for treatment
- Inform of options for diagnosis
- Inform of options for prevention
- Aggregate relevant clinical information
- Will not trigger an immediate or near term action
b) Evidenz abhängig von der Klasse nachweisen
Abhängig von der Klasse müssen die Hersteller Folgendes nachweisen:
| Grüne Klasse | Gelbe Klasse | Rote Klasse | |
|---|---|---|---|
| Analytische Validität | Ja | Ja | Ja |
| Wissenschaftliche Validität | Ja | Ja | Ja Bei neuen Verfahren zusätzliche Anforderungen |
| Klinische Leistungsfähigkeit | Nur bei neuen Verfahren | Nur bei diagnostischen Produkten der Klasse II.iii und bei neuen Verfahren | Nur bei diagnostischen Produkten und bei neuen Verfahren |
| Unabhängiges Review (Objektive und kompetente Person außerhalb des Herstellers) | Nein | Nein | Ja |
c) Notwendige Evidenz bestimmen
Falls der Hersteller über Kompetenzen und Kapazitäten verfügt, besteht der nächste Schritt darin, die notwendige Evidenz festzulegen. Das heißt, der Hersteller sollte anhand der obigen Tabelle entscheiden, welche Nachweise zu erbringen sind:
- Analytische Validität
- Wissenschaftliche Validität
- Klinische Leistungsfähigkeit
Was sich hinter diesen drei Aspekten verbrigt, finden Sie oben ausgeführt.
d) Daten erheben
Das Erheben der Daten kann genauso erfolgen wie oben beschrieben. Auch bei der FDA zählen zu den üblichen Datenquellen:
- Wissenschaftliche Fachliteratur
- Ergebnisse der Verifizierung und Validierung der Software
- Vorhandene Post-Market-Daten von eigenen und/oder fremden Äquivalenzprodukten
- Ergebnisse klinischer Studien
e) Daten auswerten und klinische Bewertung von Software erstellen
Auch bei der FDA besteht der nächste Schritt darin, die Daten auszuwerten und die klinische Bewertung bzw. (bei IVD) den abschließenden Leistungsbewertungsbericht zu schreiben. Für Medizinprodukte gibt die MEDDEV 2.7/1 Hinweise zur Struktur von klinischen Bewertungen, auch wenn das MEDDEV-Dokument die FDA nicht im Anwendungsbereich hat.
Der Auditgarant enthält neben Videos zur klinischen Bewertung auch Templates für klinische Bewertungen mit Beispielen und einer Anleitung zum Ausfüllen.
f) Fazit zu den Guidance-Dokumenten von IMDRF bzw. FDA
Der Aufwand für die klinische Bewertung bzw. Leistungsbewertung soll risikobasiert angepasst werden. Die FDA schlägt vor, bei der klinischen Bewertung von Software wie folgt vorzugehen:
Der IMDRF hat ein nicht leicht zu verdauendes Werk verfasst. Die Autoren stellen die verschiedenen Aspekte der klinischen Bewertung bzw. der Leistungsbewertung von Software gut dar. Sie verweisen auf viele weitere Publikationen der gleichen Organisation.
7. Zusammenfassung
Mit dem Dokument MDCG 2020-1 zur klinischen Bewertung bzw. zur Leistungsbewertung von Software versucht die europäische Medical Device Coordination Group den besonderen Anforderungen bei Software-Produkten Rechnung zu tragen. Die Autoren stellen die verschiedenen Aspekte der klinischen Bewertung von Software als Medizinprodukt bzw. IVD recht gut dar. Der Ansatz ist sehr viel pragmatischer als der bisherige.
Manches bleibt jedoch noch vage. Dank einiger konkreter Beispiele im Anhang wird verständlich, was gemeint ist sowie wann welcher Schritt des Algorithmus verpflichtend wird und wann nicht. Hier wäre eine genauere Abgrenzung der Notwendigkeiten (z.B. anhand der Einteilung der Funktionalitäten oder Klassifizierung der Software) sehr wünschenswert.
Außerdem fehlt jeglicher Bezug zu anderen MDCG-Guidance-Dokumenten hinsichtlich allgemeiner Aspekte der klinischen Bewertung, z.B. Anforderung an die Autoren.
Die FDA bietet ebenfalls ein Guidance-Dokument an, welches einen sehr ähnlichen Ansatz verfolgt. Hierin wird sehr viel klarer dargestellt, wann welche Nachweisausprägungen benötigt werden.
Wie bei jeder klinischen Bewertung sind die Anforderungen des MDCG-Guidance-Dokuments ohne ein wissenschaftlich und medizinisch kompetentes Team nicht umsetzbar.
Das Johner Institut ist darauf spezialisiert, klinische Bewertungen für Medizinprodukte konform mit MEDDEV 2.7/1 und MDCG 2020-1 zu erstellen und sicherzustellen, dass diese von den Benannten Stellen akzeptiert werden.
Gern unterstützt Sie das IVD-Team des Johner Instituts zudem bei der Erarbeitung einer Leistungsbewertungsstrategie für Ihre IVD-Software.
Geben Sie gerne Bescheid (z.B. über das Kontaktformular), falls Sie an der Unterstützung durch die Expertinnen und Experten des Johner Instituts für klinische Bewertungen bzw. Leistungsbewertungen interessiert sind.
Möchten Sie mehr zur klinischen Bewertung erfahren? Dann melden Sie sich gleich hier zum Bootcamp an!



Sehr geehrte Frau Dr. Martin,
vielen Dank für den sehr informativen und hilfreichen Artikel!
Ich habe eine Frage zu folgendem Abschnitt bezüglich der klinischen Leistungsfähigkeit: „Diese Daten können ebenso aus vom Hersteller durchgeführten klinischen Studien stammen wie auch aus anderen Quellen. Die Hersteller werden ermutigt, auch auf bestehende Daten von alternativen Verfahren und Produkten zurückzugreifen.“
Verstehe ich es richtig, dass für diese alternativen Verfahren/Produkte Äquivalenz NICHT nachgewiesen sein muss? Können es bspw. auch Produkte sein, die als Alternativprodukte im Rahmen von Recherchen zum Stand der Technik identifiziert wurden?
Ist die „Äquivalenzroute“ bei Software generell anwendbar oder eher nicht praktikabel?
Ich bin noch recht neu im Regulatory Affairs-Bereich und freue mich über eine Antwort!
Vielen Dank im Voraus!
Freundliche Grüße
Mareike Schenk
Sehr geehrte Frau Schenk,
vielen Dank für Ihre spannenden Fragen. Ihre erste Frage, muss ich leider verneinen. Das MDCG Dokument 2020-1 besagt: „Evidence supporting CLINICAL PERFORMANCE can be generated by testing the MDSW under evaluation, or an equivalent device, in the target population and for the intended use. The applied methodology should be appropriate in light of the device characteristics and intended purpose and may include pre-clinical testing, a clinical investigation or a clinical performance study.“
Zu Ihrer zweiten Frage ist meiner Meinung nach die Äquivalenzroute generell schwierig geworden, unabhängig ob es sich um eine Software oder ein anderes Medizinprodukt handelt. Möglich ist dies meist nur noch wenn das Äquivalenzprodukt ein Vorgängerprodukt aus dem eigenen Hause ist oder es eine vertragliche Regelung zur Offenlegung der technischen Dokumentation zwischen zwei Herstellern gibt. Meiner persönliche Meinung ist Sie für Software nicht praktikabel.
Sie können hierfür das MDGC-Dokument 2020-5 heranziehen das besagt: „Note however that the MDR specifically points out that software algorithms shall be similar in the device presumed to be equivalent. This includes software algorithms in software driving or influencing the use of a device, and in software intended to be used alone. It is the functional principle of the software algorithm, as well as the clinical performance(s) and intended purpose(s) of the software algorithm, that shall be considered when demonstrating the equivalence of a software algorithm. It is
not reasonable to demand that equivalence is demonstrated for the software code, provided it has been developed in line with international standards for safe design and validation of medical device software.“
Herzliche Grüße
Bettina Martin
Sehr geehrte Frau Dr. Martin,
vielen Dank für Ihre schnelle und ausführliche Antwort! Das hilft mir weiter. Ich werde mir das MDCG-Dokument 2020-5 nochmal genauer ansehen.
Herzliche Grüße
Mareike Schenk
Sehr geehrte Frau Dr. Martin,
vielen Dank für diese tolle Zusammenfassung der MDCG 2020-1!
Ich habe eine Nachfrage zum Verständnis. In Abschnitt 2 c) schreiben Sie:
„Manchmal sind diese Algorithmen banal. Dann stellt sich die Frage, was eine klinische Bewertung bzw. Leistungsbewertung von Software an Erkenntnissen bringen soll. In manchen Fällen kann es ausreichen, anhand von Verifizierungsergebnissen zu argumentieren.“
Was genau meinen Sie mit Verifizierungsergebnissen, bzw. könnten Sie den Aspekt nochmals erläutern?
Vielen Dank!
Viele Grüße,
Inka Benthin
Liebe Frau Benthin,
sehr gerne helfe ich Ihnen bei Ihrer Frage weiter.
Es gibt die Möglichkeit gemäß Artikel 61,10 der MDR auf klinische Daten in der klinischen Bewertung zu verzichten wenn man diese Vorgehensweise als ungeeignet erachtet (Unter bestimmten Kriterien). Bei Software niedriger Risikoklasse mit indirektem klinischen Nutzen (beispielweise bei Datenbanken, die nur gesundheitrelevante Parameter sammeln, korrekt darstellen und eventuell kalkulieren) ist es fraglich ob klinische Daten hier wirklich notwendig sind und Verifizierungsdaten ausreichen können. Dennoch muss eine vollständige klinische Bewertung mit einem ausührlichen State of the Art erstellt werden. Der Aufwand für die klinische Bewertung ist fast identisch mit dem eines Hochrisikoproduktes aufgrund der regulatorischen Forderungen.
Liebe Grüße
Bettina Martin
Liebe Frau Dr. Martin,
vielen Dank für die Erläuterung!
Das ist sehr hilfreich, da bei uns dieser Fall eine funktional getrennte Teilkomponente zutrifft. So dass wir insgesamt den kompletten Prozess durchlaufen müssen, aber in diesem Teil ggf. vereinfacht vorgehen können.
Ich wünsche Ihnen einen schöne Restwoche,
viele Grüße,
Inka benthin
Sehr geehrte Frau Martin,
eine Frage bezüglich Absatz 6 b) in dem sie die notwendigen Nachweise basierend auf der Risikoklasse des Produktes erläutern. Können Sie mir die Quelle dieser Tabelle nennen? In der FDA Guidance „Software as a Medical Device (SAMD): Clinical Evaluation“ konnte ich diese Information leider entnehmen (oder ich habe es überlesen). Oder schließen Sie dies implizit aus der Guidance für Clinical Performance Assessment für CAD devices?
Vielen Dank für ihre Hilfe.
Mit freundlichen Grüßen
A. Fink
Lieber Herr Fink,
es gibt hiefür keine Quelle. Grundlage der Tabelle ist zwar das FDA Guidance Dokument, aber wir haben diese Tabelle anhand unserer Erfahrungen und Best Practice entwickelt.
Liebe Grüße
Bettina Martin