
Inklusive kostenlosem Download der Kapitelstruktur für den klinischen Bewertungsplan
Der klinische Bewertungsplan ist eines der komplexesten Dokumente der Technischen Dokumentation. Neben dem Clinical Evaluation Report wird der Clinical Evaluation Plan (CEP) am häufigsten von den Benannten Stellen kritisiert.
Typischerweise 60 Arbeitsstunden benötigen selbst erfahrene Clinical Experts, um das oft mehr als 50-seitige Dokument so zu verfassen, dass es bei Benannten Stellen Bestand hat.
Dieser Artikel und die Kapitelstruktur zum kostenlosen Download helfen dabei,
- den klinischen Bewertungsplan schnell und gesetzeskonform zu erstellen,
- die häufigsten Fehler und damit aufwändige Nacharbeiten zu vermeiden,
- das ganze Entwicklungsprojekt zuverlässig zu planen und
- das Produkt ohne Verzögerungen und unnötigen Ärger in den Markt zu bringen.
1. Der klinische Bewertungsplan: Antworten auf die wichtigsten Fragen
1.1 Was ist ein klinischer Bewertungsplan?
Im klinischen Bewertungsplan legen Medizinproduktehersteller ihre Strategie fest, mit welchen Daten und Methoden sie die Sicherheit, die Leistungsfähigkeit und den Nutzen ihrer Produkte nachweisen und später im klinischen Bewertungsbericht dokumentieren wollen.
1.2 Gibt es eine Pflicht, einen klinischen Bewertungsplan zu erstellen?
Ja, diese Pflicht gibt es. Die MDR verpflichtet in Artikel 61 alle Medizinproduktehersteller zu einer klinischen Bewertung. Diese klinische Bewertung muss den Anforderungen des Anhangs XIV Teil A genügen. Dort heißt es:
Bei der Planung, der kontinuierlichen Durchführung und der Dokumentierung einer klinischen Bewertung haben Hersteller folgende Aufgaben: Erstellung und Aktualisierung eines Plans für die klinische Bewertung, der mindestens Folgendes enthält […]
MDR, Anhang XIV, Teil 1, Abschnitt 1
Behörden und Benannte Stellen fordern diesen klinischen Bewertungsplan konsequent ein.
1.3 Was ist der Sinn des klinischen Bewertungsplans?
Hersteller sollten den Plan aus den folgenden Gründen erstellen:
- Sie können regulatorischen Ärger vermeiden, der regelmäßig zu aufwändigen Nacharbeiten und zu einer Verzögerung bei der Inverkehrbringung der Produkte führt.
- Hersteller können dadurch rechtzeitig
- bemerken, dass ihr geplantes Produkt nicht dem Stand der Technik entspricht,
- gegensteuern, z. B. durch Anpassen der Zweckbestimmung, und
- unnötige Entwicklungsaufwände vermeiden.
- Der Plan verschafft früh Klarheit über die Dauer und Kosten des Entwicklungsprojekts. Beispielsweise entscheidet sich während dieser Planung, ob eine klinische Prüfung notwendig ist.
Sie stehen noch am Anfang Ihrer klinischen Bewertung?
Unser Clinical Evaluation Jumpstart Kit hilft Ihnen, die ersten Schritte innerhalb der klinischen Bewertung Ihrer Medizinprodukte zu gehen und verschafft Orientierung, was konkret von Ihnen verlangt wird.
1.4 Wer sollte den klinischen Bewertungsplan schreiben?
Über die notwendigen Kompetenzen verfügen Clinical oder Medical Affairs Manager oder Clinical Evaluation Specialist. Das sind Experten, die die wissenschaftlichen Methoden und das Medical Writing beherrschen.
Fehlt diese Expertise, sollten die Hersteller auf externe Dienstleister wie das Johner Institut zugreifen.
Klassische Produktmanager verfügen meist nicht über diese Kompetenzen. Selbst eine Ausbildung als Arzt oder Ärztin ist nicht hinreichend, um die Kompetenzanforderungen zu erfüllen.
2. Inhalte des klinischen Bewertungsplans
Die MDR und die MDCG-Dokumente legen die Inhalte des klinischen Bewertungsplans fest. Der Anhang XVI Teil A der MDR nennt acht Punkte (s. Abbildung 1).
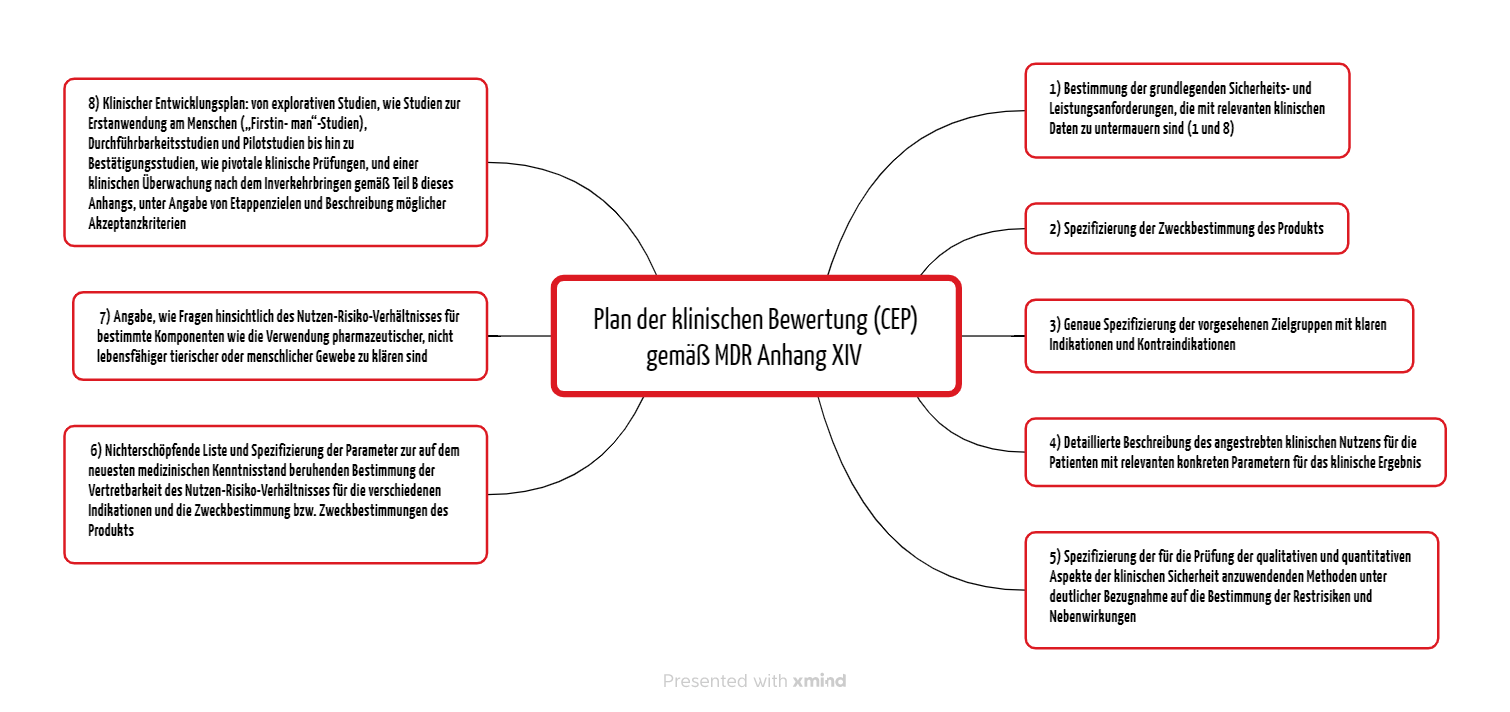
2.1 Grundlegende Sicherheits- und Leistungsanforderungen
Bereits im Clinical Evaluation Plan (CEP) müssen die Hersteller die grundlegenden Sicherheits- und Leistungsanforderungen darlegen, die sie je nach Produkt und Risiko mit relevanten klinischen Daten nachzuweisen haben. Damit erfüllen sie die Anforderungen 1 und 8 aus Annex I der MDR.
2.2 Produkt
Typischerweise erwarten die Prüfer Benannter Stellen die folgenden Informationen über das Produkt:
2.2.1 Beschreibung des Produkts
Die Beschreibung des Produkts beinhaltet:
- Eine allgemeine Beschreibung der wichtigsten Funktionselemente: seine Teile/Bestandteile (gegebenenfalls einschließlich Software), seine Formulierung, seine Zusammensetzung, seine Funktionsweise und gegebenenfalls seine qualitative und quantitative Zusammensetzung
- Die Funktionsprinzipien des Produkts und seine Wirkungsweise; Erläuterung etwaiger neuartiger Merkmale
- Bilder oder andere relevante Informationen wie Diagramme, wenn sie zum Verständnis der Verwendung des Produkts erforderlich sind
2.2.2 Klassifizierung des Produkts
Hier sollte nicht nur die Klasse, sondern auch die anwendbaren Klassifizierungsregeln und Spiegelstriche genannt werden.
2.2.3 Produktkonfigurationen/Varianten
Ebenfalls sollten die Hersteller die Produktkonfigurationen und Produktvarianten beschreiben:
- Größen, Unterschiede in den Konstruktionsmerkmalen, verschiedene Konfigurationen usw.
- Nach Möglichkeit Bilder dieser Konfigurationen und Varianten
- Beschreibung des Werdegangs und/oder der Änderungen seit der letzten Bewertung
- Gründe für die Unterschiede in den Auslegungsvarianten
2.2.4 Zubehör oder kompatible Geräte
- Alle Zubehörteile oder kompatiblen Geräte
- Im Falle eines System-/Verfahrenspakets dessen „Komponenten“
- Wenn die Verwendung von Zubehör oder kompatiblen Geräten Auswirkungen auf die klinische Sicherheit oder Leistung oder den Umfang oder die Gültigkeit der klinischen Bewertung hat, ist dies hier anzugeben.
2.2.5 Zweckbestimmung
- Die vorgesehene Patientenpopulation und die medizinischen Bedingungen, die diagnostiziert, behandelt und/oder überwacht werden
- Indikationen und Kontraindikationen
- Detaillierte Beschreibung des angestrebten klinischen Nutzens für die Patienten mit relevanten und konkreten Outcome-Parametern für das klinische Ergebnis. Dieser Punkt ist essenziell, um die klinische Zulassungsstrategie für das Produkt festzulegen. Hierbei müssen die Hersteller allerdings schon den Stand der Technik kennen (s. Abschnitt 2.3).
Diese Elemente sollten wörtlich aus dem Dokument Zweckbestimmung oder der Produktbeschreibung übernommen werden und in allen Dokumenten der Technischen Dokumentation identisch sein (beispielsweise auch in der Gebrauchsanweisung). Hierauf achten die Benannten Stellen sehr.
2.3 Stand der Technik
Hersteller sind verpflichtet, den Stand der Technik zu ihrem Produkt zu ermitteln, um die richtige klinische Strategie wählen zu können. Die systematische Literaturrecherche wird benötigt, um ihr Produkt im Vergleich zum Stand der Technik zu bewerten.
Deswegen evaluieren die Clinical Experts des Johner Instituts diesen Stand der Technik gleich zu Beginn der Entwicklungsprojekte.
Der Stand der Technik hilft auch bei der Entscheidung, ob für die klinische Bewertung des Medizinprodukts klinische Daten benötigt werden (ggf. durch eine klinische Prüfung) oder ob Leistungsdaten ausreichend sind.
Der Fachartikel zur Literatursuche sowie der Medical-Writer-Kurs helfen bei einer zielgerichteten Suche und bei einem präzisen Formulieren einer gesetzeskonformen klinischen Bewertung.
2.4 Klinischer Nutzen mit klinischen Parametern und Akzeptanzkriterien
Damit können die Hersteller klinisch relevante Parameter mit Akzeptanzkriterien bestimmten, um den klinischen Nutzen nachzuweisen. Es ist hilfreich, sich dazu in den medizinischen Kenntnisstand einzuarbeiten und zu analysieren, welche Risiken und Nebenwirkungen dabei entstehen können.
Ein Kühlpack soll der Schmerzlinderung dienen. Der Hersteller bestimmt als Parameter die erreichbare Hauttemperatur und als Akzeptanzkriterium Werte kleiner 13 °C.
Diese Vorüberlegungen sind nützlich, um
- die Komplexität des Projekts besser zu verstehen,
- das Projektteam zielgerichtet mit Informationen zu unterstützen,
- abzuwägen, ob man bestimmte Claims überhaupt machen kann,
- zu entscheiden, welche risikominimierenden Maßnahmen getroffen werden müssen,
- bei Bedarf klinische Endpunkte klinischer Studien zu bestimmen.
Bei Bestandsprodukten ist hier zu überprüfen, ob sich der Stand der Technik geändert hat und die vorhandene Zweckbestimmung und der Nutzen so noch gehalten werden können.
Bei diesem Schritt kann sich herausstellen, dass klinische Parameter keinen Sinn ergeben oder nicht existieren und sich die Sicherheit, Leistung und Nutzen besser mit technischen Parametern nachweisen lässt.
Die Parameter und die Akzeptanzkriterien müssen dem Stand der Technik entsprechen.
Im Fachartikel zu den klinischen Endpunkten erfahren Sie, weshalb die klinischen Endpunkte so relevant sind und wie Sie diese in klinischen Prüfungen nachweisen.
2.5 Datenroute und klinische Strategie
2.5.1 Die Alternativen
Nachdem bekannt ist, was nachgewiesen werden muss, gilt es festzulegen, wie die Nachweise erbracht werden, dass das Produkt dem Stand der Technik entspricht.
Es gibt drei wesentliche Strategien, um diese Nachweise bei der klinischen Bewertung zu erbringen (s. Abb. 2):
- Zuerst gibt es eine Unterscheidung zwischen klinischen Daten und Leistungsdaten (3), dann eine
- Unterscheidung, wie die klinischen Daten erhoben werden, nämlich mit dem eigenen Produkt (1)
- oder einem Äquivalenzprodukt (2).
Bei den klinischen Daten zum eigenen Produkt lässt sich noch unterscheiden, ob eine klinische Prüfung notwendig ist oder nicht. Letzteres gilt, wenn es ausreichend klinische Daten aus PMS- und PMCF-Aktivitäten gibt.
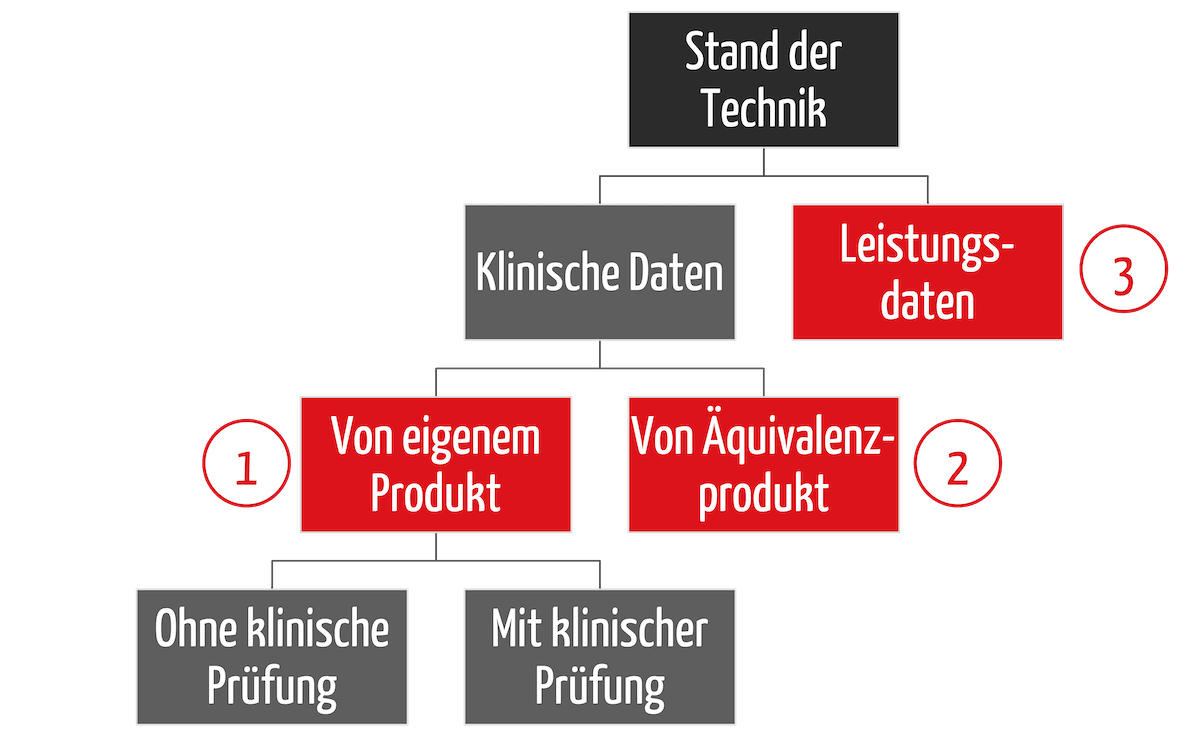
Die Strategie, den Nachweis nur über Leistungsdaten zu führen (3), die auch „Data Route“ genannt wird, wird bereits im Clinical Evaluation Plan festgelegt. Viele Benannte Stellen fordern zusätzlich einen Bewertungsplan (Appraisal Plan). Dieser beschreibt, wie welche Art von Daten bewertet werden.
Falls Sie die Äquivalenzroute in Betracht ziehen, ist es ratsam, den Äquivalenzvergleich bereits im CEP durchzuführen und im Appraisal Plan zu beschreiben, anhand welcher Daten und Kriterien Sie ein Produkt als ausreichend äquivalent betrachten.
Der Fachartikel zur Äquivalenz beschreibt detailliert, wie diese Äquivalenznachweise gelingen.
Bei der Wahl der „Datenroute“ sind die Hersteller nicht frei. Mehrere regulatorische Anforderungen sind zu beachten (s. Tabelle 1).
2.5.2 Die regulatorischen Anforderungen
| MDR | Datenroute der klinischen Bewertung |
| Anhang VII, Kapitel III | Ist das Medizinprodukt ein implantierbares Medizinprodukt oder ein Klasse-III-Medizinprodukt? |
| Artikel 61, 6b | Handelt es sich um Nahtmaterial, Klammern, Zahnfüllungen, Zahnspangen, Zahnkronen, Schrauben, Keile, Zahn- bzw. Knochenplatten, Drähte, Stifte, Klemmen oder Verbindungsstücke? |
| MDCG-2020-6 | Ist das Medizinprodukt ein existierendes unter der MDD/AIMDD (= Bestandsprodukt)? |
| Wurde das betreffende Produkt durch Änderungen eines bereits von demselben Hersteller in Verkehr gebrachten Produkts konzipiert? | |
| Anhang XIV, Teil A, 3 | Gibt es ein Medizinprodukt, dessen technische, biologische und klinische Merkmale in einer Weise gleichartig mit dem evaluierenden Medizinprodukt sind, dass es keinen klinisch bedeutsamen Unterschied bei der Sicherheit und klinischen Leistung der Produkte gibt? |
| Artikel 2, 48 | Gibt es klinische Daten zum Medizinprodukt aus folgenden Quellen: (1) klinische Prüfung(en) des betreffenden Produkts, (2) in nach dem Peer-Review-Verfahren überprüfter wissenschaftlicher Fachliteratur veröffentlichte Berichte über sonstige klinische Erfahrungen mit dem betreffenden Produkt, (3) klinisch relevante Angaben aus der Überwachung nach dem Inverkehrbringen, insbesondere aus der klinischen Nachbeobachtung nach dem Inverkehrbringen? |
| Artikel 2, 51 | Hat das Medizinprodukt klinische Endpunkte und hat es klinische Aussagen (Claims)? |
| Artikel 61, 10 | Wird der Nachweis der Übereinstimmung mit grundlegenden Sicherheits- und Leistungsanforderungen auf der Grundlage klinischer Daten für ungeeignet erachtet? |
Legen Sie die klinische Strategie bzw. Datenroute nicht erst im klinischen Bewertungsbericht (Clinical Evaluation Report) fest, sondern bereits im klinischen Bewertungsplan (Clinical Evaluation Plan).
2.6 Methoden zur Untersuchung der klinischen Sicherheit und Leistungsfähigkeit
Hersteller müssen ausführen, wie die Nachweise zu führen sind, und auch die Methoden nennen. Möglich sind etwa:
- Usability Tests
- Stoß- und Falltests
- Prüfung der elektromagnetischen Sicherheit
- Materialcharakterisierungen
- Software-Integrationstests
- Penetration-Tests
- Tierversuche
- Dauertests
- Klinische Prüfungen
- PMS-Daten
- PMCF-Daten
Dabei sollten die Hersteller die angewendeten Normen benennen.
2.7 Klinische Evidenz
Die MDR verpflichtet Hersteller in Artikel 61, die Güte und den Umfang der Daten festzulegen. Sie nennt das den „klinischen Nachweis“.
Die klinischen Daten und die Ergebnisse der klinischen Bewertung zu einem Produkt, die in quantitativer und qualitativer Hinsicht ausreichend sind, um qualifiziert beurteilen zu können, ob das Produkt sicher ist und den angestrebten klinischen Nutzen bei bestimmungsgemäßer Verwendung nach Angabe des Herstellers erreicht;
Der englische Begriff clinical evidence umschreibt besser, worum es geht.
Die notwendige klinische Evidenz hängt wiederum vom Produkt, seinem Risiko und vom Stand der Technik ab.
Handelt es sich um ein Nischenprodukt, kann eine Studie mit einer kleinen Patientenpopulation vollkommen angemessen sein, während bei einem anderen Produkt, die Fallzahl je nach Risikoklasse nicht ausreichend ist.
Auch diese klinische Evidenz sollten die Hersteller im Clinical Evaluation Plan bestimmen.
Die klinische Evidenz, die gewählte Datenroute und die optionale Beschreibung einer klinischen Prüfung sind Teil des sogenannten klinischen Entwicklungsplans.
2.8 Appraisal Plan
Zudem fordert die MEDDEV 2.7.1 rev.4 aus dem Jahr 2016 im Abschnitt 9.2 einen „Appraisal Plan“.
Dieser Plan sollte nicht nur die Literaturbewertung berücksichtigen. Auch weitere Daten sind systematisch zu bewerten, beispielsweise:
- Test-Reports
- PMS-Daten
- Gebrauchsanweisungen
- Marketingmaterialien
3. Klinischer Bewertungsplan: Die Kapitelstruktur
Weder die Vorgaben der MDR (Anhang XIV) noch das MDCG 2020-6 für Bestandsprodukte (Anhang II) erzwingen eine Kapitelstruktur. Sie sollte jedoch so gewählt sein, dass sie den Leser vom Allgemeinen ins Spezielle führt.
Einen Vorschlag für solch eine Kapitelstruktur finden Sie hier zum Download.
4. Der klinische Bewertungsplan im Entwicklungsprozess
Der klinische Bewertungsplan ist ein Dokument, das die Hersteller sehr früh im Entwicklungsprozess erstellen sollten; denn als Teil dieses Plans bestimmen sie den Stand der Technik. Dieser „State of the Art“ muss bei weiteren Dokumenten und Inhalten bekannt sein und berücksichtigt werden (s. Tabelle 2).
| Dokument, Inhalte | Bemerkungen |
| Zweckbestimmung | Die Zweckbestimmung muss dem Stand der Technik entsprechen. Umgekehrt folgen aus der Zweckbestimmung auch die Claims, die nachgewiesen werden müssen. Der klinische Bewertungsplan legt fest, wie diese Nachweise geführt werden. |
| Klinisch relevante Parameter mit Akzeptanzkriterien | Aus diesen Parametern und Akzeptanzkriterien folgen meist Produktanforderungen. |
| Vorläufige Gefährdungsanalyse | Daraus können risikominimierende Maßnahmen bzw. Anforderungen an das Produkt folgen. |
| Notwendigkeit und Design der klinischen Prüfung | Die klinische Bewertung muss beispielsweise überprüfen, ob die o. g. Akzeptanzkriterien erreicht werden. Die Endpunkte dieser Studien müssen dem Stand der Technik entsprechen. |
Die Informationen beeinflussen einander (s. Abbildung 3).
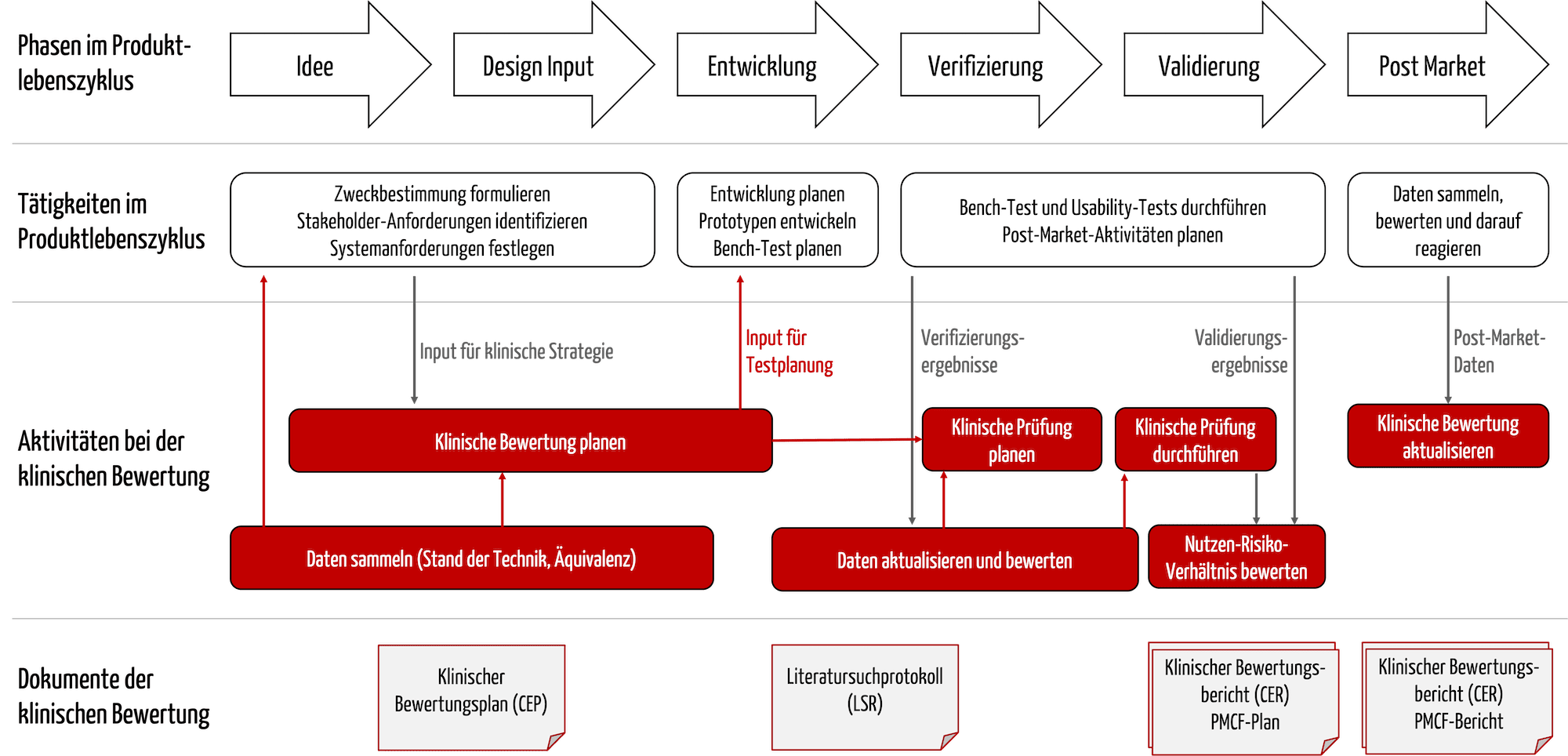
Unternehmen sollten darauf achten und die Prozesse so gestalten, dass die Clinical Affairs Experts von Beginn an in die Formulierung der Zweckbestimmung involviert sind. Andernfalls drohen Rücksprünge und unnötige Aufwände und Verzögerungen.
5. Typische Fehler erkennen und beheben
| Fehler | Behebung |
| Der Hersteller erklärt nicht sein Produkt, sondern referenziert nur andere Dokumente. | Sowohl der CEP als auch CER enthalten diese Informationen (leider) redundant, z. B. Zweckbestimmung, Indikationen, Kontraindikation sind identisch mit den anderen Dokumenten. |
| Der klinische Nutzen fehlt. | Der Hersteller spezifiziert klinische Claims und quantifiziert diese mit Parametern (s.o.). |
| Der Hersteller nennt für die Parameter keine Akzeptanzkriterien. | Für jeden dieser Parameter legt der Hersteller einen oder mehrere Akzeptanzkriterien fest. |
| Der Appraisal Plan fehlt komplett oder nicht ausführlich genug. | Der Hersteller sollte mit der Benannten Stelle abklären, ob diese einen „Appraisal Plan“ verlangt und falls ja, in welcher Form. |
| Die klinische Strategie ist nicht eindeutig. | Der Hersteller beschreibt genau, wie er den Nachweis von Sicherheit, Leistungsfähigkeit und Nutzen führt. Dazu legt er u. a. fest, ob dies anhand klinischer Daten erfolgt und ob diese Daten von einem eigenen Produkt oder einem Äquivalenzprodukt stammen. |
| Der Plan ist nicht auf Bestandsprodukte angepasst (siehe MDCG-2020-6). | Auch der klinische Bewertungsplan muss aktualisiert werden. Beispielsweise kann die klinische Strategie bei Bestandsprodukten mit PMS- und PMCF-Daten sowie ausreichender klinischer Evidenz abgedeckt werden. |
| Der Hersteller beginnt die Planung der klinischen Bewertung nach der finalen Formulierung der Zweckbestimmung oder sogar erst während der Verifizierung des Produkts. | Der klinische Bewerter legt die klinische Strategie bereits während der Formulierung der Zweckbestimmung fest. Falls das nicht erfolgt, können in der Zweckbestimmung klinische Claims stehen, die später mit einer klinischen Studie bewiesen werden müssten, obwohl man vielleicht einen anderen Weg über Leistungsdaten hätte einschlagen können. |
| Der Hersteller kommuniziert mit seiner Benannten Stelle erst, nachdem die klinische Bewertung verfasst ist. | Insbesondere bei Neuprodukten mit innovativen Aspekten ist es ratsam, sich schon im Vorfeld mit der Benannten Stellen auszutauschen, um sicherzustellen, dass diese mit der gewählten klinischen Strategie mitgeht. Benannte Stellen dürfen zwar nicht beraten tätig sein, aber sie dürfen Auskunft geben, ob die klinische Strategie sinnvoll ist. Diese Rückfragen beim Clinical Reviewer der Benannten Stelle empfehlen sich auch vor dem Beseitigen von kritisierten Mängeln. |
Beachten Sie auch den Fachartikel zu den 5 häufigsten Irrtümern und Fehlern bei klinischen Bewertungen. Er gibt Tipps, wie Sie diese Fehler vermeiden können.
6. Zusammenfassung und Fazit
6.1 Eine anspruchsvolle Aufgabe
Einen klinischen Bewertungsplan zu erstellen, ist eine der anspruchsvollsten Aufgaben:
- Die Kompetenzanforderungen sind sehr hoch. Das betrifft die wissenschaftliche Kompetenz ebenso wie die notwendigen Kenntnisse des Produkts und des medizinischen Kontexts.
- Das Dokument ist sehr umfangreich und „inhaltsschwer“. Die Daten zusammenzutragen ist sehr aufwändig. Selbst erfahrene Clinical Affairs Experts benötigen dafür ein bis zwei Wochen.
- Die Erstellung setzt eine enge und intakte Interaktion mit fast allen Unternehmensbereichen voraus und wirkt sich auf den ganzen Produktlebenszyklus aus.
- Es scheint keine Einigkeit bei den Benannten Stellen zu geben, was die Anforderungen an diesen Plan betrifft. Die Anforderungen unterscheiden sich nicht nur zwischen den Benannten Stellen, sondern auch innerhalb dieser. Auch das führt zu regelmäßigen Abweichungsberichten.
Selbst wenn die Aufgabe schwierig und aufwändig ist, sollten Hersteller diese nicht aufschieben. Denn mit dem Verfassen eines validen und präzisen klinischen Bewertungsplans haben sie bereits wegweisende Festlegungen getroffen.
6.2 Wegweisende Festlegungen
Mit dem Verfassen des klinischen Bewertungsplans haben die Hersteller …
- sichergestellt, dass das zu entwickelnde Produkt dem Stand der Technik entsprechen wird.
- Risiken identifiziert und Maßnahmen bzw. Produktanforderungen abgeleitet.
- die notwendigen Methoden für die Produktprüfung festgelegt.
- Klarheit erreicht, ob eine klinische Prüfung notwendig ist (eine wesentliche Voraussetzung für die Projektplanung).
- die Grundlagen für einen konformen klinischen Bewertungsbericht und damit die Zulassung des Produkts gelegt.
6.3 Das A und O der klinischen Bewertung
Der klinische Bewertungsplan ist eines der ersten Dokumente, mit dem die Hersteller beginnen sollten. Er hat Auswirkungen auf andere Dokumente wie die Zweckbestimmung und den Design-Input.
Umgekehrt ist der klinische Bewertungsbericht eines der letzten Dokumente, das der Hersteller abschließt, weil es sich auf fast alle anderen Dokumente der Technischen Dokumentation bezieht.
In anderen Worten: Der klinische Bewertungsplan ist das Alpha und der klinische Bewertungsbericht das Omega nicht nur der klinischen Bewertung, sondern der ganzen Produktentwicklung.
Beide Dokumente müssen fortlaufend aktualisiert werden.
Die Clinical Experts des Johner Instituts helfen Ihnen dabei, Ihren klinischen Bewertungsplan in kürzester Zeit so zu erstellen, dass Ihre Benannte Stelle ihn durchwinken wird. Dann haben Sie eine solide Basis, u. a. auch für Ihren klinischen Bewertungsbericht.
Mit dem Bewertungsplan verfügen Sie implizit auch über Ihre klinische Strategie.
- Sie stellen sicher, dass Ihr geplantes Produkt dem Stand der Technik entspricht und vermarktet werden darf.
- Sie kennen die Risiken Ihres Produkts und können zu einem frühen Zeitpunkt Maßnahmen festlegen, was schneller und günstiger ist, als dies später zu tun.
- Sie wissen, welche Nachweise Sie mit welchen Methoden erbringen müssen. Das hilft Ihnen, die Aufwände zu schätzen und einen realistischen Projektplan zu erstellen.
- Sie können rechtzeitig prä-klinische Tests oder eine klinische Prüfung beauftragen und damit den Zeitplan einhalten und sich Stress ersparen.
Mininieren Sie Ihre (nicht nur) regulatorischen Risiken und nutzen Sie die Hilfe der Clinical Experts des Johner Instituts. Melden Sie sich gleich.



Vielen Dank für diesen sehr wichtigen Beitrag!
Meine Frage bezieht sich auf den Stand der Technik: gemäß IMDRF/GRRP WG/N47 definiert sich dieser aus den konsolidierten Erkenntnissen aus Wissenschaft, Technologie UND Erfahrung. Würden Sie alternative, neuartige therapeutische Optionen, zu denen es noch wenig klinische Erfahrungswerte gibt, in diesem Kapitel trotzdem beschreiben?
Auch wenn diese noch nicht den aktuellen Stand der Technik widerspiegeln, macht es ja Sinn, diese im Rahmen der klinische Nachbeobachtung nach dem Inverkehrbringen zu beobachten.
Vielen Dank und liebe Grüße!
Lieber Michael,
vielen Dank für deinen Kommentar und dein Interesse an diesem wichtigen Thema.
Die Berücksichtigung solcher Therapien, zu denen es noch wenig klinische Erfahrung gibt, ist tatsächlich ein wichtiger Aspekt. Es ist sinnvoll, diese Innovationen zu erkunden, um den medizinischen Fortschritt zu fördern, allerdings müssen ihre Sicherheit und Wirksamkeit gründlich bewertet werden. Dein Vorschlag, diese Therapien im Rahmen der klinischen Nachbeobachtung nach der Markteinführung zu überwachen, ist sehr wertvoll. Dieser Ansatz ermöglicht es, notwendige Daten zu sammeln, ohne den etablierten Stand der Technik vorschnell zu erweitern.
Um Innovation zu fördern und gleichzeitig Patientensicherheit zu gewährleisten, ist es ratsam, solche Therapien in einem separaten Abschnitt zu diskutieren, der sich mit zukünftigen oder in Entwicklung befindlichen Optionen befasst. Dies fördert Transparenz und ermutigt zu weiterer Forschung und Entwicklung.
Liebe Grüße!