
An das Labeling stellen sowohl die europäischen als auch die US-amerikanischen Regularien Anforderungen. Allerdings verstehen beide Rechtssysteme den Begriff nicht völlig identisch. Selbst die Schreibweisen unterscheiden sich: Labeling in den USA, Labelling in Europa.
Erfahren Sie in diesem Artikel, was Sie beim Labeling jeweils beachten müssen. Apropos Schreibeweise: Wir verwenden in diesem Artikel das amerikanische Englisch und sparen uns ein „l“ :-).
Labeling: Was ist das?
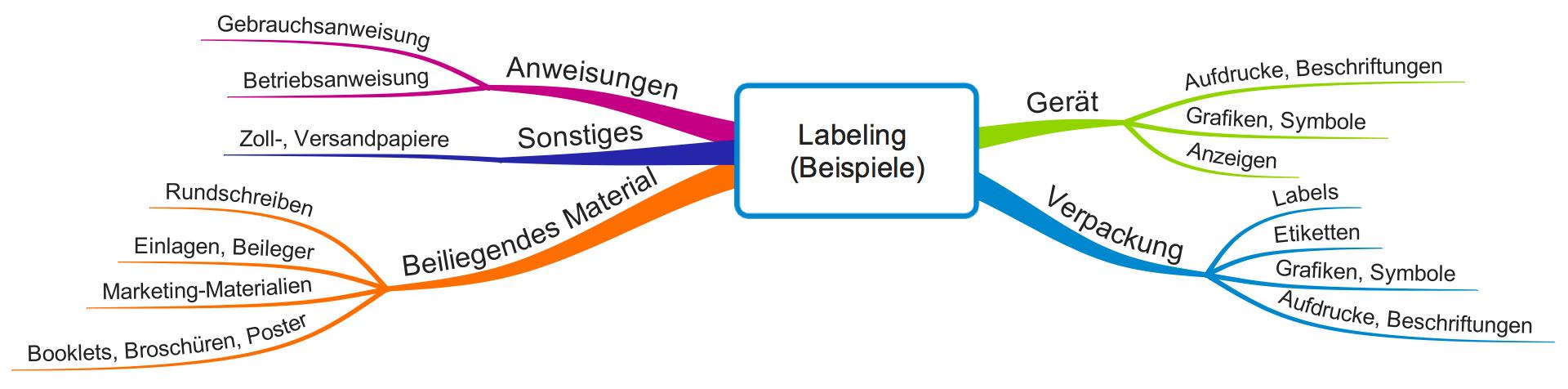
Abbildung 1 verschafft Ihnen einen Überblick über Artefakte, die verschiedene Regularien zum Labeling zählen.
a) Definition (FDA)
Eine Definition liefert die FDA:
„all labels and other written, printed, or graphic matter
- upon any article or any of its containers or wrappers, or
- accompanying such article“ at any time while a device is held for sale after shipment or delivery for shipment in interstate commerce.
Dabei sind ‚label’ laut FDA:
display of written, printed, or graphic matter upon the immediate container of any article […]
Die FDA nennt als Beispiele für das Labeling:
- Betriebs- bzw. Gebrauchsanweisungen
- Aufdruck auf Verpackungen (z.B. Grafiken, Symbole, Texte)
- Etiketten
- Flyer, Booklets, Broschüren, Poster
- Einlagen, Beileger
- Rundschreiben
D.h. auch „begleitendes“ Marketing-Material zählt zum Labeling. Den Begriff „accompanying“ möchte die FDA wörtlich verstanden wissen. Selbst Zoll- und Versandpapiere können unter die Definition fallen.
b) Definition (ISO 13485)
Die Begriffsdefinition der ISO 13485 hat sich im Lauf der Zeit leicht geändert. In der Version 2012 hieß es noch:
jede geschriebene, gedruckte oder graphische Information
- auf einem Medizinprodukt oder einem seiner Behältnisse oder sonstigen Verpackungen, oder
- als Beilage zum Medizinprodukt,
die sich auf die Identifizierung, technische Beschreibung und Verwendung des Medizinprodukts bezieht; ausgenommen sind Versanddokumente.
Die Übersetzung des Begriffs „Labelling“ war somit „Kennzeichnung“. Im Gegensatz zur FDA fallen die Versanddokumente nicht unter den Begriff.
In der neusten Version der Norm, der ISO 13485:2016, heißt es:
label, instructions for use, and any other information that is related to identification, technical description, intended purpose and proper use of the medical device, but excluding shipping documents
Diese Definition verwendet den Begriff „Label“. Diesen definiert die Norm nicht.
c) Medizinprodukteverordnung
Die Medizinprodukteverordnung (MDR) verwendet den Begriff „Labelling“, ohne ihn zu definieren. Folgende Textstellen lassen vermuten, dass die MDR das Labeling eher als die Beschriftungen direkt am Gerät versteht:
- „In the labelling, instructions for use, making available, putting into service and advertising of devices, it shall be, […]“
- „the instructions for use and the labelling“
Offensichtlich unterscheidet die MDR Gebrauchsanweisungen und Marketingmaterial einerseits und „Labelling“ andererseits.
d) Sonstige Regularien
In diesem Kontext verwendet die IEC 60601-1 die Begriffe „Kennzeichnung“, „Aufschriften“ und „Unterlagen“. Im Englischen „Identification“, „Marking“ und „Documents“.
e) Fazit
Unterschiedliche Regularien haben zwar vergleichbare, aber nicht übereinstimmende Definitionen des Begriffs Labeling / Kennzeichnung. Stellen Sie also sicher, dass Sie, Ihre Kolleginnen und Kollegen, Ihre Kunden und Ihre benannte Stelle vom Gleichen sprechen.
Regulatorische Anforderungen an das Labeling
Übersicht
Die MDR geht davon aus, dass die Kennzeichnung auf dem Produkt selbst (Etikettierung) und die Gebrauchsanweisung existieren und die in Artikel 23, Anhang I aufgeführten Informationen enthalten. Weitere Regularien stellen konkretere Anforderungen:
- Die EU-Verordnung 2226/2021 regelt die Verwendung elektronischer Gebrauchsanweisungen.
- Die ISO 24971 empfiehlt im informativen Anhang, die Etikettierung bei der Risikoanalyse zu berücksichtigen.
- Die ISO 13485:2016 fordert, dass die „Kennzeichnung“ Teil der Medizinprodukteakte ist. Bei der Produktion muss überwacht werden, dass die Kennzeichnung und Verpackung den Vorgaben entsprechen.
- Die IEC 60601-1 sowie darin referenzierte Normen wie die DIN EN 60445 und die DIN EN 60447 geben zahlreiche Regeln, über die Sie sich weiter unten einen Überblick verschaffen können.
- Weitere Partikularnormen der IEC 60601-Familie ergänzen diese Vorgaben.
- Die ISO 15223-1 regelt die Symbole, die Hersteller von Medizinprodukten zu verwenden haben.
- Die ISO 20417 ergänzt und präsisiert Anforderunderungen der MDR und IVDR ans Labeling.
Beispiele für Anforderungen der IEC 60601
Die IEC 60601-1 widmet dem Thema Labeling gleich ein ganzes Kapitel. Darin geht es um die Verwendung von Symbolen und Farben, um geforderte Aufschriften, Gebrauchsanweisungen, um die dauerhafte Lesbarkeit usw. usw.
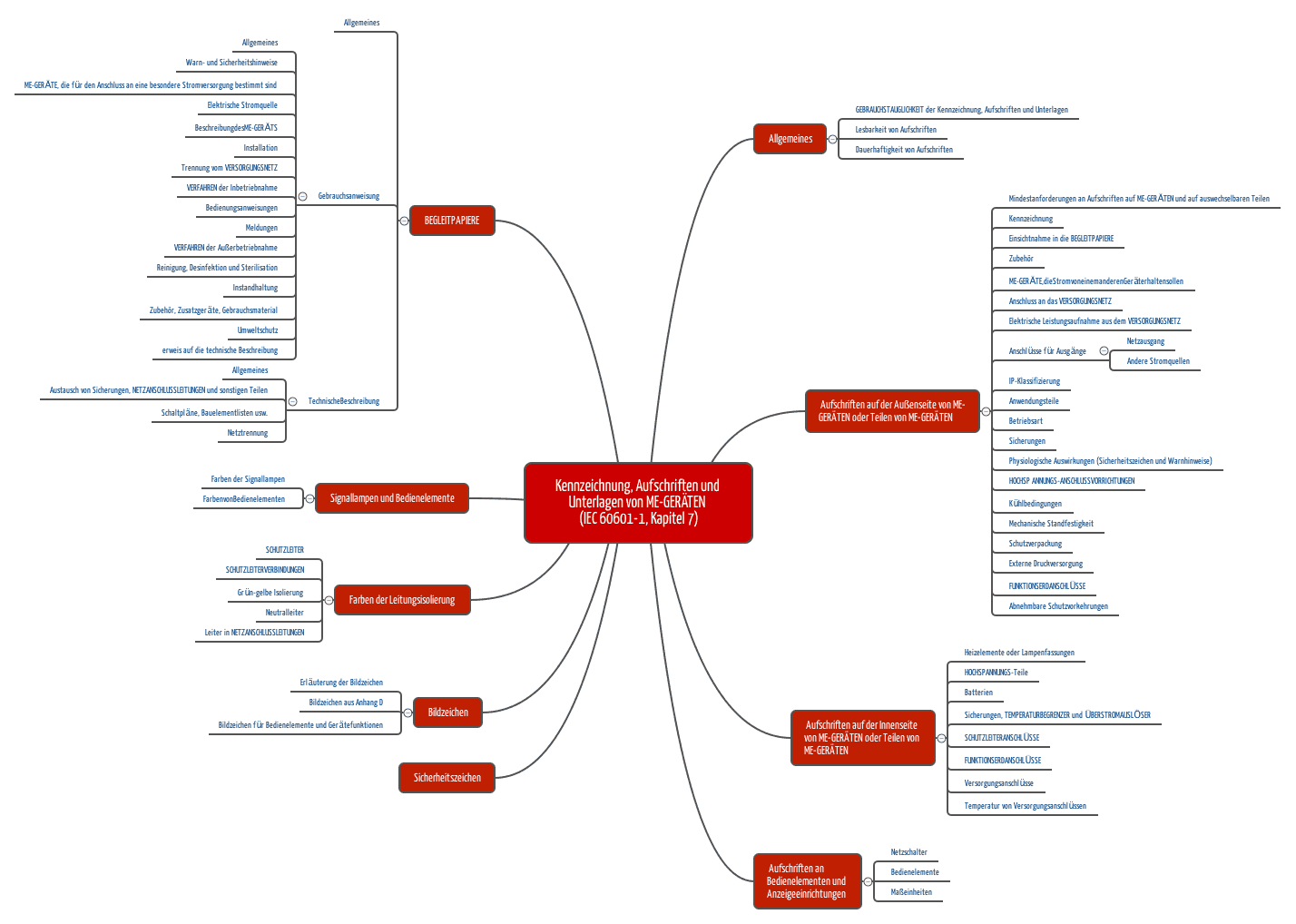
(zum Vergrößern klicken)
Die wichtigsten Anforderungen beziehen sich somit auf die:
- Aufschriften an der Außenseite des Geräts
- Aufschriften an der Innenseite des Geräts
- Aufschriften an Bedienelementen und Anzeigen
- Gebrauchsanweisung
- technische Beschreibung und
- sonstigen Aspekte wie Farben, Symbole und Signallampen.
Zusammenfassung
Das Labeling spielt eine wichtige Rolle, um Medizinprodukte sicher und wirkungsvoll für Patienten, Anwender und Dritte zu nutzen. Daher müssen die Hersteller verstehen, welche Risiken dadurch minimiert und welche dadurch ggf. geschaffen werden. Dabei müssen sie zahlreiche Regularien beachten.



Folgende Frage: Gibt es regulatorische Anforderungen, aus denen hervorgeht, wie ein Label auf der Verpackung korrigiert werden kann?
Beispiel:
Auf der Verpackung (Karton) ist ein Fehler im aufgedruckten UDI (sowohl im AIDC und HRI).
Könnte man mit einem Aufkleber das fehlerhafte UDI mit dem korrekten „einfach“ überkleben?
Oder müsste das fehlerhafte Label unleserlich gemacht werden bevor ein Korrektur-Label aufgeklebt wird?
Oder kann man die Verpackung nicht verwenden und sie muss neu produziert/gedruckt werden?
Vielen Dank im Voraus für Ideen
Sehr geehrter Herr Kress,
mir sind keine expliziten Anforderungen für den Austausch bzw. die Korrektur von Labels bekannt. Daher sehe ich auch keine Einschränkungen bei der Art dieser Korrektur außer der Maßgabe, dass dies eine Änderung am Produkt ist, die u.a. im Risikomanagement und bei den weiteren grundlegenden Anforderungen betrachtet werden muss und ggf. eine erneute Verifizierung und Validierung nach sich zieht.
Wenn das Label beispielsweise im „Design“ geändert würde, wären ggf. Usability Evaluationen notwendig. Wenn das Label überklebt würde, müsste beispielsweise geprüft werden, dass dieses Label im Rahmen des bestimmungsgemäßen Gebrauchs – d.h. auch bei Transport und Lagerung – weiter stabil und lesbar bleibt. Wenn es eine Änderung am AIDC ist, wären ggf. erneute Prüfungen entsprechend der Normen wie ISO 15415ff. in Betracht gezogen werden.
Falls all diese Anforderungen erfüllt sind und erfüllt bleiben, sind Sie frei bei der Wahl der Änderung. Achten Sie aber auf ggf. erneute Freigaben.
Beste Grüße, Christian Johner
Bei der Verwendung von Label Templates welche Symbole und Texte dynamisch auf Typenschildern / Verpackungslabels usw. Produkt- & länderspezifisch füllen bzw. deren Sichtbarkeit steuern – wie muss da die technische Dokumentation zum einzelnen Produkt aussehen?
Reicht ein 100% Label aller verwendeten Labelvorlagen mit Platzhaltern ink. Beschreibung wann wie welche Symbole in welchem Kontext ausgeprägt werden sowie die Version und Änderungshistorie dieser verwendeten Labels oder muss jeweils ein eine produktspezifisches „Vorschau“ des Labels hinterlegt werden?
Letzteres kann ich quasi nur automatisiert versuchen zu erzeugen da ich sonst händisch fortlaufend 100erte Dokumentation von Hand anfassen müsste.
Dankeschön & Grüße
Sehr geehrter Herr Kraut,
danke für Ihre anspruchsvolle Frage!
Da ich nicht ganz sicher bin, ob ich diese ausreichend verstehe: Stehen Sie vor der Aufgabe, dass Sie in der technischen Dokumentation für sehr, sehr viele Medizinprodukte angeben wollen, wie die Labels jeweils gestaltet sind?
Es gibt generell keine Vorgabe, ob Sie das Label grafisch oder wie von Ihnen vorgeschlagen als eine Kombination aus grafischer und textueller Darstellung beschreiben. Die Beschreibung muss letztlich eindeutig sein, die zugehörige Dokumentation gelenkt.
Falls sich besondere Aspekte der Usability ergeben, müssten Sie diese untersuchen.
Beste Grüße, Christian Johner
Vielen Dank für die Antwort und verzeihen sie das Delay.
„Stehen Sie vor der Aufgabe, dass Sie in der technischen Dokumentation für sehr, sehr viele Medizinprodukte angeben wollen, wie die Labels jeweils gestaltet sind?“
So kann man es in der Tat zusammenfassen.
Die Produkte selbst sind mit ihren Attributen selbst im PLM / ERP gepflegt und Änderungen selbstverständlich dokumentiert. Genauso haben die wenigen Labelvorlagen für den Druckprozess hier eine Versionierung mit Historie.
Beim Druck ziehen diese Daten dynamisch aus PLM und ERP an und erzeugen das konkrete Label, welches auf dem Drucker landet.
Diese Labelvorlage verhält sich dabei wie eine Schraube die in jedem Produkt verbaut ist – damit eine einheitliches Design und weniger Aufwand bei beim Label-Design anfällt, single point of change eben.
Dieser Vorteil der hohen Wiederverwendung hebt sich allerdings mit den betriebenen Aufwendungen auf, bei einer (kleinsten) Änderung an der Label Vorlage im worst case die gesamte Produktpalette versionieren zu müssen + die technische Dokumentation auf Stand zu bringen. (so sieht es der Prozess aktuell hier vor)
Wenn bei der Dokumentation im jeweiligen Produkt lediglich der Verweis auf die aktuellste Label-Vorlage samt ihrer Historie und der prinzipiellen Beschreibung wie die Ausprägung des Labels zu Stande kommt ausreicht, wäre das von Vorteil, da ich nicht bei 1000 Produkten aufwendig einen Label-Validierung fahren muss um diese wieder in der technischen Dokumentation zu aktualisiere. Dann ist es über einen Verweis geregelt.
Mit anderen Worten, ich validiere die Label Vorlage bei jeder Änderung (schaue ob z.B. alle Symbole passen, Texte da landen wo sie sollen), nicht aber jedes aus dieser Vorlage entstandene Label. Aktuell wird dieses quasi als digitale Grafik in die Technische Doku eingespielt, manuell.
(in der Produktion wird die Qualität des Labels natürlich unabh. davon nochmals sichergestellt)
das ist wirklich eine spannende weil grundlegende Fragestellung in Bezug auf effiziente moderne datenbasierte Dokumentation bzw. datenbasierter Variantensteuerung.
Erst einmal ist dabei natürlich die Frage zu beantworten, ob die Datenquellen (ERP, PIM, Redaktionelle Inhalte) geprüft und freigegeben sind. Dann stellt sich die Frage, welchen Einfluss kann die technische Beschaffenheit des Templates und dessen Varianzmechanik auf die individuellen, dynamisch erzeugten Outputs haben. Ich nehme an. dass man bei einer Änderung eines Label-Templates nicht ausschließen kann, dass irgendein damit erzeugtes Label-Derivat dadurch fälschlicherweise beeinflusst werden kann. Wenn damit ein unvertretbares Risiko verbunden ist (Label falsch) muss eine Gegenmaßnahme her. Neben einer erneuten Prüfung aller 1000 Label-Varianten bleibt dann eigentlich nur die 100% Prüfung des Label-Inhalts in der Herstellung.
Ggf. kann ich mir theoretisch einen automatischen Prozess vorstellen, der nach jeder Template-Anpassung alle Labels rausproduziert und diese auf „optische“ Veränderungen eines Bildes/PDFs im Vergleich zum ursprünglich freigegebenen Master prüft und mir dann alle Abweichungen meldet.
Das Thema lässt sich auf diverse andere Dinge übertragen, die mitunter viel aufwändiger sind, als es der optische Abgleich eines Labels ist.
– Digital und on-Demand bereitgestelltes Begleitmaterial aus einer Single-Source-Quelle
– Die Firmware einer gemeinsamen Komponente für eine neue Produktvariante konfigurieren.
Unterm Strich scheint mir in vielen fällen „Template-basierte“ Variantensteuerung irgendwie nicht lohnend zu sein, wenn damit eine Prüfung unzähliger Outputs nach jeder Änderung des Templates verbunden ist.
Je weitreichender (=effizienter) ein Template eingesetzt wird, desto aufwändiger ist jede Änderung eben dieses Templates, wenn man das nicht automatisiert bekommt.
Sehr geehrter Herr Dr. Johner,
eine Frage stellt sich mir zur Zulässigkeit von Angaben auf der Verpackung, die, wenn man aus dem Bereich der Regulatorischen Betreuung von Arzneimitteln kommt, werblichen Charakter haben. Sind diese durch die MDR oder andere Regularien verboten (wie es im Arzneimittelbereich der fall ist)? Jegliche Werbung für Medizinprodukte muss auch bspw. dem HWG entsprechen, aber gibt es im Medizinprodukterecht gesonderte Anforderungen zur Werbung?
Vielen Dank im Voraus und freundliche Grüße
Irene Carlson
Sehr geehrte Frau Carlson,
bei den Medizinprodukten ist die Sache etwas anders, und sie wird auch nicht durch das Medizinprodukterecht alleine geregelt.
Was verboten ist, sind natürlich jegliche irreführenden Angaben. Alle Angaben, die Sie mit Bezug zur Sicherheit, Leistungsfähigkeit und Wirksamkeit des Medizinprodukts machen, müssen Sie im Rahmen der klinischen Bewertung nachweisen. Allerdings gibt es auch „Marketing-Claims“, die nicht darunterfallen.
Beste Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
ist der Inverkehrbringer eines Medizinproduktes juristisch gesehen die selbe Person wie der Hersteller? Die Frage zielt darauf ab, ob der Inverkehrbringer eines Medizinproduktes auf seinem Label auch das Symbol für Hersteller (Fabriksymbol) vermerken muss oder nicht (im Falle eines Händlers, ist das Symbol z.B. nicht auf dem Etikett).
Vielen Dank im Voraus und viele Grüße
Georg Bilow
Sehr geehrter Herr Bilow,
der Inverkehrbringer und der Hersteller können, müssen aber nicht notwendigerweise die gleiche Person sein. Sie finden die Definition im Artikel zur Inverkehrbringung.
Wie Sie sehen hängt das auch davon ab, ob der Hersteller in der EU sitzt.
Das Fabriksymbol wird für den Hersteller benötigt. Falls dieser nicht auch der Inverkehrbringer ist, weil er nicht in der EU sitzt, so muss der Bevollmächtigte auf der Kennzeichnung angegeben werden.
Die MDR fordert in Anhang I zur Kennzeichnung:
Beste Grüße, Christian Johner
Hallo Herr Prof. Dr. Johner,
ist Ihnen eine Norm etc. bekannt die vorgibt oder daraufhin weißt welche Abzugskräfte ein Typenschild (z.B. Warnhinweis/ auf einem Kunststoffgehäuse) auf einem Medizinprodukt der Klasse 2 erfüllen muss. Oder gibt es diesbezüglich keine Anforderungen/Vorgaben. Bei einem internen Audit sind wir auf diese Thematik gestoßen und diskutieren nun über die vorhandene Sachlage.
Vielen Dank im Voraus und viele Grüße
Patrick Beuerle
Sehr geehrter Herr Beuerle,
danke für die Frage!
Eine Norm ist mir nicht bekannt. Diese Abzugskräfte hängen wie alle grundlegenden Sicherheits- und Leistungsanforderungen nicht von der Klassifizierung ab. Vielmehr sollten diese abhängig von dem Risiko und der Nutzungsumgebung festgelegt werden.
In der IEC 60601-1 gibt es aber Hinweise, wie man die Dauerhaftigkeit von Beschriftungen prüfen soll.
Viele Grüße, Christian Johner
Guten Tag Hr. Prof. Johner,
ich habe eine Frage zur Etikettierung (Typenschilder und Verpackungsetiketten). Gibt es eine Vorgabe in welchen Sprachen die Produktbeschreibung auf dem Etikett erfolgen muss? Wir haben bisher DE/ EN/ FR/ IT/ ES auf unseren Etiketten. Sind weitere Sprachen Pflicht, wenn wir auch in andere EU-Länder liefern?
Für Gebrauchsanweisungen gibt es da ja eine Vorgabe aber für die Etikettierung habe ich bisher nichts finden können.
Danke für Ihre Rückmeldung.
Sehr geehrte Frau Lerch,
als Experte fürs Labelling beim Johner Institut beantworte ich gerne Ihre Frage.
In welcher Sprache die Etikettierung (Label) vorliegen muss, ist in der nationalen Gesetzgebung des jeweiligen Landes geregelt. Grundsätzlich muss das Label wie die Gebrauchsanweisung auch in der jeweiligen Landessprache des Landes, in dem das Produkt vertrieben wird, vorliegen (siehe MDR Artikel 10 Absatz 11: https://www.johner-institut.de/blog/wp-content/uploads/2020/02/MDR_konsolidiert.html#artikel-10). In Sonderfällen, z.B. wenn die vorgesehenen Nutzer professionelle Benutzer sind, sind landesspezifische Ausnahmen möglich.
Herzliche Grüße
Nils Becker
Sehr geehrter Herr Professor Johner,
ich habe eine Frage zur CE Kennzeichnung auf Transportverpackungen (Verpackung der OP- Tische für die Spedition).
Durch die MDR ist eindeutig geklärt, dass der UDI- Code auf dem Produkt selbst und den Handelsverpackungen (ausgenommen sind Transportverpackungen/ Versandcontainer) aufgedruckt sein muss.
Nun stellt sich mir die Frage, wie es sich mit der Anbringung des CE Zeichens verhält.
Auf dem Label des Produktes und der Handelsverpackung ist der Aufdruck des CE Zeichens im Artikel 20 eindeutig festgelegt. Jedoch finde ich keinen Hinweis, ob die Transportverpackung/ Versandcontainer, wie beim UDI- Code, auch hier ausgenommen ist oder nicht.
Ist Ihnen evtl. eine andere Norm bekannt, in der es, bezüglich Transportverpackung und CE Kennzeichnung, eine eindeutige Festlegung gibt?
Vielen Dank vorab und herzliche Grüße
Karoline Mair
Sehr geehrte Frau Mair,
gerne antworte ich Ihnen an Stelle von Herrn Johner.
Sie haben recht, das CE-Kennzeichen muss auf dem Produkt selbst bzw. seiner Primärverpackung und Handelsverpackung angegeben werden. Darüber hinaus ist mir keine regulatorische Anforderung bekannt, die besagt, dass das CE-Kennzeichen auf Transport- oder Versandcontainern angebracht werden muss. Es gibt aber auch keine Bestimmung, die dagegen spricht.
Ich hoffe meine Antwort hilft Ihnen weiter, ansonsten fragen Sie gerne nach.
Herzliche Grüße
Nils Becker
Sehr geehrter Herr Professor Johner,
ich habe eine Frage bezüglich der Verwendung ablösbarer Klebeetiketten auf Verpackungen zur Kennzeichnung des Produkts mit den geforderten Angaben.
Zunehmend fordern unsere Kunden(SH), dass wir unsere Produktverpackungen mit abziehbaren Etiketten versehen.
Auf den Etiketten befinden sich nun alle erforderliche Informationen gemäß MDR.
Wenn die SHs diese Etiketten nun für ihre eigene Dokumentation ablösen, sind die Produkte dann noch MDR-konform gekennzeichnet ?
Nach meiner Einschätzung nicht. Wie stehen Sie dazu?
Vielen Dank im Voraus!
Mit freundlichen Grüßen
Christine Graß
Liebe Frau Graß,
da liegen sie vollkommen richtig. Artikel 10(11) der MDR schreibt explizit, dass „Die Angaben auf der Kennzeichnung (müssen) unauslöschlich, gut lesbar und für den vorgesehenen Anwender oder Patienten klar verständlich sein“ müssen. Um dies zu gewährleistet, wäre ein zusätzliches Abziehlabel denkbar.
Liebe Grüße
Philipp Schleer
Sehr geehrter Herr Schleer,
vielen Dank für Ihre zeitnahe Antwort und Bestätigung meiner Einschätzung.
Mit freundlichen Grüßen
Christine Graß
Sehr geehrter Herr Prof. Johner!
Bei einem internen Meeting mit unserem Team sind wir auf eine interessante Marketing-Idee gestoßen, wollen uns aber rechtlich noch absichern.
Ist es möglich, dass man auf ein Medizinprodukt der Klasse IIb (Elektisches Therapiegerät) auf der Vorderseite einen neuen Namen platziert, wobei auf der Rückseite der Original-Name und alle relevanten Daten bestehen bleiben? Wir haben einen Medizinproduktehandel, sind in diesem Fall nur Händler, nicht Inverkehrbringer des Produktes.
Vielen Dank!
Liebe Grüße, Karl R-W
Lieber Karl R-W,
prinzipiell ist dieses Vorgehen nicht unmöglich. Artikel 16 schreibt hierzu: „(1) Ein Händler, Importeur oder eine sonstige natürliche oder juristische Person hat die Pflichten des Herstellers bei Ausführung folgender Tätigkeiten: a) Bereitstellung eines Produkts auf dem Markt unter dem eigenen Namen, dem eigenen eingetragenen Handelsnamen oder der eigenen eingetragenen Handelsmarke, außer in den Fällen, in denen ein Händler oder Importeur eine Vereinbarung mit einem Hersteller schließt, wonach der Hersteller als solcher auf der Kennzeichnung angegeben wird und für die Einhaltung der nach dieser Verordnung für die Hersteller geltenden Anforderungen verantwortlich ist;“. Demzufolge ist die beschriebene Vereinbarung zwischen Hersteller und Händler bei einem derartigen Vorgehen essentiell und ich würde Ihnen raten für die Überprüfung einen Medizinprodukteanwalt hinzuzuziehen, um sich rechtlich abzusichern.
Liebe Grüße
Philipp Schleer
Sehr geehrter Herr Professor Johner,
Artikel 20 (5) EU-MDR „CE-Konformitätskennzeichnung“ gibt an. „[…] Diese Kennnummer ist auch auf jeglichem Werbematerial anzugeben, in dem darauf hingewiesen wird, dass das Produkt den Anforderungen für die CE-Kennzeichnung erfüllt.“ Leitet sich hieraus (oder auch an anderer Stelle der MDR) eine Verpflichtung ab, dass Marketingmaterial CE zu kennzeichnen ist?
Vielen Dank im Voraus und freundliche Grüße,
Dr. Martina Zell
Sehr geehrte Frau Dr. Zell,
auf Marketingmaterialien muss nicht zwingend eine CE-Kennzeichen angebracht werden. Sollten Sie allerdings eines anbringen, dann immer mit der dazugehörigen Kennnummer, insofern eine Benannte Stelle involviert ist.
Liebe Grüße
Philipp Schleer
Sehr geehrter Herr Professor Johner,
es geht um die Angaben auf dem Etikett, die die Identifizierung des Produkts ermöglichen. Eine dieser Angaben wäre die Artikelnummer, richtig?
Außerdem geht es um die Verwendung von Symbolen auf dem Etikett.
Ist es Pflicht ,das sog. REF-Symbol vor die Artikelnummer zu schreiben? Oder kann die Artikelnummer auch ohne dieses Symbol auf das Etikett aufgebracht werden?
Vielen dank für Ihre Hilfe!
Mit freundlichen Grüßen
Christine Graß
Lieber Herr Graß,
gemäß der ISO 20417:2021 muss eine dedizierte Produktidentifikation gegeben sein, dies kann die Modellnummer oder Artikelnummer sein oder auch der Handelsname des Produkts sein. Die Artikelnummer kann dabei entweder als Symbol oder in Form von Text als solche gekennzeichnet sein. Die MDR schreibt hierzu in Anhang I 23.2 g) „g) die Losnummer oder die Seriennummer des Produkts nach dem Wort „LOSNUMMER“ oder „SERIENNUMMER“ oder gegebenenfalls einem gleichwertigen Symbol;“ Demzufolge können Sie auch anstelle des REF-Symbols einen schriftlichen Hinweis, bspw. „ARTIKELNUMMER“, geben, solange für den Anwender erkenntlich ist, um welche Nummer es sich handelt.
Liebe Grüße
Philipp Schleer
Sehr geehrter Herr Professor Johner,
nach Artikel 20 der MDR wird davon gesprochen, dass die CE-Kennzeichnung auf das Produkt, der Verpackung und der Handelsverpackung aufgebracht werden muss. Soweit so gut.
Wir habe eine Handelsverpackung, in der Produkte der Klasse I aber auch der Klasse II enthalten sind. Welches CE muss dann dort aufgebracht werden? Muss ich auf die Kennzeichnung evtl. eine „Packliste“ mit Artikelnummern und der entsprechenden CE-Kennzeichnung aufbringen?
Vielen Dank
Mit freundlichen Grüßen
Guido Herr
Sehr geehrter Herr Herr,
besten Dank für Ihre Frage!
Wenn Sie zwei Produkte unterschiedlicher Klassen in einer Handelsverpackung haben, vermute ich, dass es sich um eine Behandlungseinheit handelt.
In dem Fall müssten Sie nach MDR Artikel 22 gar kein CE-Kennzeichen auf der Handelsverpakung anbringen (Vorausgesetzt sie ändern die Zweckbestimmung der Produkte nicht. Dann müssten Sie ein neues Konformitätsbewertungsvefahren durchlaufen.).
Ich hoffe, meine Antwort hilft Ihnen weiter.
Herzliche Grüße
Nils Becker
Sehr geehrter Herr Professor Johner
Wir stehen vor der Frage, in welcher Sprache die Produktbezeichnung bei einem Klasse IIb-Implantat auf dem Patientenetikett, welches zum Implantationsausweis mitgeliefert wird, ausgestellt werden muss?
Muss auch hier, wie bei der Produktverpackung auf die Mehrsprachigkeit/Landessprache geachtet werden, oder reicht es, die Produktbezeichnung auf z.B. Englisch anzugeben.
Ich hoffe Sie können helfen, diese Wissenslücke zu schliessen.
Beste Grüsse aus der Schweiz
Jürgen Kaufman
Sehr geehrter Herr Kaufmann,
vielen Dank für Ihre Frage. Ich antworte gerne an Herrn Johners Stelle.
Die Kennzeichnung auf dem Produkt selbst („Etikett“) bzw. die Kennzeichnung auf der Verpackung muss in der vom Mitgliedstaat (in welchem das Produkt Inverkehr gebracht wird) geforderten Sprache(n) bereitgestellt werden (siehe MDR Artikel 10 Absatz 11). Artikel 18 stellt die gleichen Sprachanforderungen für Informationen, die dem Implantat mitgeliefert werden sowie für den Implantationsausweis. Demnach müssen „Angaben zur Identifizierung des Produkts einschließlich des Produktnamens“ in der jeweils geforderten Sprache gegeben werden.
Ich hoffe meine Antwort hilft Ihnen weiter.
Herzliche Grüße
Nils Becker
Hallo Herr Professor Johner,
generell gilt ja die Regel, dass die vollständige Kennzeichnung auch auf der nächst höheren Verpackung angebracht wird, wenn es erheblichen Platzmangel auf der Kennzeichnung des Produkts an sich gibt.
Wir haben IVD-Produkte mit erheblichem Platzmangel.
Gibt es hier eine Best Practice, in welcher Reihenfolge die Informationen auf der Kennzeichnung des Produkts bei Platzmangel angebracht werden sollte?
Z.B. man befüllt den vorhandenen Platz zuerst mit der Ref. Nr., dann Lot/SN Nr., dann Hersteller , …. bis nichts Weiteres mehr drauf passt und dann natürlich alle notwendigen Informationen auf der nächst höheren Verpackungskennzeichnung.
Wir sind in einer internen Diskussion, welche Informationen Priorität erhalten.
Herzlichen Dank vorab und viele Grüße,
Corinna Bausch
Liebe Frau Bausch,
vielen Dank für Ihre Frage. Gerne antworte ich an Herrn Johners Stelle.
Die ISO 18113 Teile 1-5 geben konkrete Vorgaben zu Ihrer Fragestellung. Der Primärbehälter sollte folgende Angaben enthalten:
– 6.2 Hersteller
– 6.3.1 Name des IVD-Reagenz oder Bestandteils
– 6.3.2 Chargenbezeichnung
– 6.4 Inhaltsangabe
– 6.5 Gebrauch für die In-vitro-Diagnostik
– 6.6 Lagerungs- und Handhabungsbedingungen
– 6.7 Verfallsdatum
– 6.8 Warnhinweise und Vorsichtsmaßnahmen
Bei Platzmangel können laut Kapitel 6.1.2 folgende Angaben gekürzt oder ausgeklammert werden: 6.4, 6.5 und 6.6. Sollte der Platz für die restlichen Angaben immer noch nicht ausreichen, empfehle ich risikobasiert zu entscheiden, welche Angaben auf dem Primärbehälter ausgeklammert werden können.
Ich hoffe, ich konnte Ihnen weiterhelfen.
Herzliche Grüße
Nils Becker
Sehr geehrter Hr. Johner,
wir haben eine Rückfrage zur Kennzeichnung des Herstelldatums bei sterilen MP. Lassen sich die Angaben in den Abschnitt 23.2 +3 (i), (j) des Anhang I in Kombination mit Abschnitt 23.3 (h) der MDR so interpretieren, dass es auch bei sterilen Produkten erlaubt ist, das Herstelldatum ausschließlich als Bestandteil des Barcodes anzuzeigen?
Grüße
Hartmut Simon
Sehr geehrter Herr Simon,
vielen Dank für Ihre Frage. Für den UDI-Träger findet sich die Antwort in Anhang VI, Teil C, Artikel 3.5 der MDR:
„3.5. Wird auf der Kennzeichnung eine Losnummer, eine Seriennummer, eine Software-Identifikation oder ein Verfallsdatum angegeben, so ist diese bzw. dieses Teil der UDI-PI. Befindet sich auf der Kennzeichnung auch das Herstellungsdatum, so muss dieses nicht in die UDI-PI aufgenommen werden. Befindet sich auf der Kennzeichnung nur das Herstellungsdatum, so ist dieses als UDI-PI zu verwenden.“
Man muss also nur zwingend das Haltbarkeitsdatum von sterilen Produkten kodieren, das Herstelldatum ist optional.
Herzliche Grüße,
Nils Becker
Sehr geehrter Prof. Johner,
ist es bei einer reinen Software als Medical Device notwendig, das Herstellungsdatum mit auf dem Splashscreen anzugeben? Und falls ja, wie ist dieses Datum dann definiert?
Vielen Dank im Voraus für Ihre Antwort,
mit freundlichen Grüßen,
Frank Feyerabend
Sehr geehrter Herr Feyerabend,
auch für Software ist die Angabe eines „Herstelldatums“ erforderlich – hierfür bietet sich das Release Datum der jeweiligen Version an. Neben dem Herstelldatum sollten Sie auch die Versionsnummer und die daraus abgeleitete UDI (inklusive UDI-PI) mit angeben. Denn das müssen Sie (spätestens nach Ablauf von Übergangsfristen) auch tun.
Der Splash Screen ist eine perfekte Möglichkeit, um der Pflicht zur Kennzeichnung nachzukommen.
Beste Grüße, Christian Johner
Lieber Johner Institut Team,
ich habe eine Frage zu Verpackungslabeln.
Wir haben ein Medizinprodukt, dem ein Zubehörteil, welches selbst als Medizinprodukt definiert ist, in der Handelsverpackung begelegt werden soll. Diese Zubehörteile sind in kleine Tütchen verpackt, die auch ein entprechendes Labeling enthalten (GA und Verpackungsabel). Muss das Verpackungslabel der Zubehörteile auch auf die Handelsverpackung des eigentlichen Produktes, so dass ersichtlich ist, dass die Verpackung noch zusätzliche Medizinprodukte enthält? Oder reicht es die Zubehörteile innerhalb der Verpackung zu Kennzeichnen und die Handlesverpackung enthält nur das Label des Basisprodukts.
Leider konnte ich hier in den Regularien dazu finden. Es heißt meist, das das Produkt entsprechend gekennzeichnet sein muss, entweder selbst oder auf der Verpackung. Lediglich Artikel 20 der MDR lässt darauf schließen, dass die Verpackung ein CE Kennzeichen enthalten muss, dieses wäre schon drauf, da es sich bei beiden Produkten um Klasse I Produkte handelt.
Vielen Dank und freundliche Grüße
Eva Esslinger
Sehr geehrte Frau Esslinger,
hier ist die Frage, wie Sie ihr Basisprodukt in Verkehr bringen. Geschieht das ausschließlich zusammen mit dem Zubehör, dann wäre das Zubehör nur eine Komponente und es müsste außen nicht explizit aufgezählt werden, da Sie ein nur ein Produkt auf den Markt bringen. Natürlich müssen Sie Warnungen und Vorsichtsmaßnahmen, die das Zubehör betreffen, auch nach außen so kommunizieren, beispielsweise wenn das Zubehör anfälliger gegenüber Umweltbedingungen sein sollte oder eine geringere Haltbarkeit aufweisen sollte.
Sind aber nun beides eigenständige, vollständig getrennte Medizinprodukte (also Basisprodukt ohne Zubehör sowie das Zubehör selbst), dann hätten Sie ein System nach Artikel 22 und müssten eine entsprechende Erklärung abgeben. Sie müssten dann auch die Kennzeichnung beider Produkte strikt trennen, also Basisprodukt / Zubehör / System – davon gehe ich aber von Ihrer Beschreibung her nicht aus.
Mit besten Grüßen
Christopher Seib
Sehr geehrter Herr Johner,
unser Medizinprodukt (SaMD) ist eine Library, welche verschiedene Berechnungen zur Verfügung stellt und in eine andere SW eingebunden und von dieser verwendet werden kann.
Wir stellen auch einen Splash-Screen via API zur Verfügung, mit dem wir der Kennzeichnungspflicht (Hersteller, Herstelldatum, Versionsnummer, UDI, …) nachkommen, dieser muss aber von der SW die unsere Library verwendet, explizit aufgerufen werden. Außer dem Spash-Screen hat unsere SW kein GUI.
Frage: Da unsere SaMD keine klassische Verpackung und Installation hat (nur Download), stellt sich die Frage, ob wir beim Download auch die Informationen zur Kennzeichnung zur Verfügung stellen müssen (z.B. in einem eigenen txt-File) und ob bei der Installation/Download der SW, die unser SaMD nutzt, die Kennzeichnung unseres Medizinproduktes ersichtlich sein muss.
Mit besten Grüßen
Gerald Kohlmaier
Sehr geehrter Herr Kohlmaier,
da Sie über den Splash Screen alle nötigen Informationen zusammen mit Ihrem Produkt liefern, sind die Anforderungen an die Kennzeichnung nach Anhang I, Artikel 23.1 der MDR ausreichend erfüllt.
Eine zusätzliche .txt Datei wäre hier nicht erforderlich.
Mit besten Grüßen
Christopher Seib
Sehr geehrters Johner Team,
Welche Angaben müssen auf einem Shipping Container gemacht werden welcher verschiedene Arten von Medizinprodukten enthält? (Basiseinheit + Zubehör) Die Medizinprodukte innerhalb des Containers haben ihre eigene Verpackung und entsprechendes Labeling nach ISO 20417. Soweit ich informiert bin muss auf den Shipping Container kein CE Zeichen, ebenso sind Shipping Container von der UDI ausgeklammert, wie verhält es sich nun mit den restlichen Angaben wie Artikelnummer/ Serialnummer/ Produktnamen/ Lagerungskonditionen?
Muss von außen ersichtlich sein welche Produkte sich in dem Shipping Container befinden?
Leider konnte ich keine Guidance hierzu finden sowohl für EU als auch für USA.
Ich hoffe Sie können mir hier weiterhelfen 🙂
Vielen Dank!
Viele Grüße,
Marcel Wagner
Sehr geehrter Herr Wagner,
der Shipping Container ist von der Kennzeichnungspflicht unter der MDR ausgenommen – das betrifft wie Sie richtig festgestellt haben auch die UDI. Die Rückverfolgbarkeit (über die Seriennummer etc.) ist auf Ebene der Versandcontainer nicht gefordert. Auf dem Container sollten Sie aber aus Gründen der Produktsicherheit zumindest die Informationen festhalten, die einen Einfluss auf die Stabilität nehmen könnten, z.B. die Lagertemperatur. Zusätzlich treffen hier ggf. weitere Regularien, welche den Transport von Gefahrstoffen betreffen, zu.
Herzliche Grüße
Christopher Seib
Sehr geehrters Johner Team,
wir haben ein Medizinprodukt welches auch ein Gefahrstoff ist und daher auch nach der Verordnung (EG) Nr. 1272/2008 gekennzeichnet werden muss. Nun müssen wir Änderungen der Kennzeichnung bzgl. der Verordnung (EG) Nr. 1272/2008 vornehmen, hat das Auswirkungen auf die Verifizierung ect. bzgl. des Medizinproduktes?
Lieber Steffan,
gemäß Anhang I, Artikel 23.2 (m), der MDR müssen Warnhinweise auf der Kennzeichnung angegeben werden und stellen somit einen regulatorischen Inhalt dar. Müssen nun Änderungen vorgenommen werden – sei es nun, dass diese sich wie in diesem Fall aus einer weiteren Verordnung oder aber aus anderen Gründen ergeben – so muss der Hersteller dies im Rahmen seiner Verifizierung und Validierung risikobasiert betrachten und es sollte eine Neubewertung der Sicherheit erfolgen. Ist die Änderung signifikant, muss gegebenenfalls muss auch die benannte Stelle informiert werden.
Herzliche Grüße
Christopher Seib
Vielen Dank Christopher,
gibt es einen Leitfaden bei welchen Änderungen die benannte Stelle informiert werden muss.
Vielen Dank.
Lieber Steffan,
Eine Entscheidungshilfe der MDCG zu signifikanten Änderungen findet sich unter: https://health.ec.europa.eu/system/files/2020-09/md_mdcg_guidance_significant_changes_annexes_en_0.pdf
Herzliche Grüße
Christopher Seib
Sehr geehrtes Team,
Muss auf dem Primärocontainer eines Medizin Produkts der Klasse IIa (elektr. Absaugpumpe) das Verfallsdatum eines im Lieferumfang beiliegenden Schlauchs (Nicht steril) gekennzeichnet sein?
Vielen Dank.
Mit freundlichen Grüßen,
Ralf Gambel
Sehr geehrter Herr Gambel,
das ist korrekt, sofern beides zusammen verpackt und vertrieben wird! Der Anwender muss direkt von außen erkennen können, wie lange das Produkt sicher zu lagern und zu verwenden ist. Hier sollten sie immer nur das geringste Haltbarkeitsdatum aller enthaltenen Produkte angeben, wenn also Ihr Schlauch bis 2024 haltbar ist und die Absaugpumpe bis 2028, sollte sich im Haltbarkeitsdatum nur die 2024 wiederfinden. Es macht hier auch keinen Unterschied, ob sie die beiden Produkte als System oder als zwei Komponenten desselben Produktes in Verkehr bringen.
Herzliche Grüße
Christopher Seib
Sehr geehrtes Team vom Johner Institut,
unserer Frage bezieht sich beim Labeling auf folgendes. In ganz vielen Gebrauchsanweisungen für Medizinprodukte findet man die Typenschilder oder die Hauptkennzeichnung des Produktes in einem Kapitel wieder. Können Sie uns beantworten aus welchen regulatorischen Anforderungen dieser Punkt kommt? Weder in der MDR noch in der ISO 20417 oder der ISO 9687 sowie der ISO 15223-1 findet sich darauf ein konkreter Hinweis, dass die direkte Produktkennzeichnung in der Gebrauchsanweisung auftauchen muss. Der Inhalt und der Abgleich natürlich schon.
Eine direkter Abdruck der Typenschilder oder Produktkennzeichnung für bei Änderungen zu ungewollten Redundanzen oder Fehlern in den Begleitpapieren, wenn diese nicht nachgezogen werden.
Viele Grüße und danke für die Rückmeldung
Ralph Hoffmann
Sehr geehrter Herr Hoffmann,
es gibt generell keine strikte regulatorische Vorgabe, dass Sie ein Etikett in die Gebrauchsanweisung aufnehmen müssen. Einige Hersteller tun dies, um die Forderung aus der IEC 60601 zu erfüllen (Begleitpapiere müssen unter anderem die Modell- oder Typbezeichnung enthalten) und um die Gebrauchsanweisung einem spezifischen Produkt besser zuordnen zu könne; es verpflichtet Sie jedoch nichts dazu, diese Anforderungen (sofern bei Ihnen überhaupt anwendbar) über ein abgedrucktes Typenschild zu erfüllen.
Herzliche Grüße
Christopher Seib
Sehr geehrtes Team des Johner Instituts,
wir optimieren gerade unser Produktlabeling und möchten für ein Klasse 1 Produkt ein sogenanntes Leporello-Etikett verwenden, welches direkt obig am Produkt befestigt ist.
Mit dieser neuen Lösung ist die Herstelleradresse sowohl im Inneren des Etiketts verfügbar als auch auf dem „Base Label“, welches unterhalb des Leporello-Etiketts auf dem Produkt zu finden ist. Dies würde bedeuten, dass die Herstelleradresse auf dem Produkt vorhanden, aber nicht direkt sofort sichtbar wäre. Man muss also erst das Etikett „öffnen“, um die Herstelleradresse zu sehen. Ist dies MDR-Konform?
Leider konnten wir dahingehend keine Informationen finden, ob eine sofortige Sichtbarkeit eine MDR-Anforderung darstellt oder nicht.
Artikel 10 (11) spricht „nur“ von „Die Angaben auf der Kennzeichnung müssen unauslöschlich, gut lesbar und für den vorgesehenen Anwender oder Patienten klar verständlich sein“.
Anhang 1 23.1b) spezifiziert lediglich, dass „Die für die Kennzeichnung vorgeschriebenen Angaben werden auf dem Produkt selbst angebracht“
Besten Dank für Ihre Rückmeldung.
MFG
Dirk Schäfer
Sehr geehrter Herr Schäfer,
der Begriff „deutlich lesbar“ ist in der ISO 20417 etwas genauer erklärt, in Anhang B finden Sie einen Prozess zur Überprüfung. Es sollten alle Anforderungen aus der vorgesehenen Position des Anwenders eindeutig zu erkennen sein. Grundlegend würde ein Verdecken von Informationen auch der ISO 14971 und der IEC 62366 widersprechen. Alle Informationen zur Produktsicherheit und Rückverfolgbarkeit sollten dem Anwender zur Verfügung stehen, ohne dass das Produkt dazu geöffnet werden muss – das Entfernen eines Etikettes ist bereits als Öffnen zu bewerten, da die Originalverpackung beschädigt wird.
Herzliche Grüße
Christopher Seib
Sehr geehrtes Team vom Johner Institut,
die GSPR 10.4.1 besagt, dass Medizinprodukte CMR-Stoffe nur dann in einer Konzentration von mehr als 0,1% Massenanteil enthalten dürfen, wenn dies entsprechend gerechtfertigt ist.
Bezieht sich der 0,1% Massenanteil auf die Komponente des Medizinprodukts, welche den CMR Stoff enthält oder auf das Gewicht des gesamten Medizinprodukts?
Meine Frage daher, muss das Medizinprodukt auch dann entsprechend mit dem CMR-Symbol gekennzeichnet werden, wenn nur eine Komponente mehr als 0,1% Massenanteil des CMR Stoffs enthält, die Konzentration des CMR-Stoffs auf das gesamte Medizinprodukt berechnet aber unter 0,1% Massenanteil liegt?
Vielen Dank für Ihre Einschätzung!
Schöne Grüße,
Martina M.
Liebe Martina,
entscheidend ist hier der Zweck dieses Symbols: Der Hersteller muss und möchte den Anwender gemäß der eigenen Risikoanalyse warnen, dass eine Gefährdung besteht, wenn er mit dem Produkt in Kontakt kommt. Berührt der Anwender eine hochgradig krebserregende oder toxische Substanz, ohne vorab über deren Vorhandensein informiert worden zu sein, so bringt ihn das möglicherweise in Lebensgefahr. Ist diese Substanz im gesamten Produkt gleichmäßig verteilt, so ist das Risiko eher gering. Tritt der entsprechende Stoff an eine Stelle gehäuft auf, so ist der Kontakt hier deutlich gefährlicher.
Daher bezieht sich die MDR auch auf den Massenanteil. Dieser stellt per Definition den Anteil einer Substanz in einem Gemisch dar. Entsprechend ist der CMR-Anteil pro Substanz / Komponente zu bewerten und nicht über das gesamte Produkt. Liegt er an einer Stelle über 0,1%, so ist eine entsprechende Warnung möglichst frühzeitig anzugeben, also beispielsweise auf der Verpackung.
Ich hoffe, das beantwortet die Frage!
Herzliche Grüße
Christopher Seib
Dear Dr. Johner,
I am writing regardin the query on the contents of the medical devcie label, as PMDA mentions that labeling requirement shall be fullfilled as per ISO 13485 and ISO 13485 does not enlist the required data (contents) for the labels but is only mentions the labeling definition that is taken from GHTF/SG1/N70:2011. Although, GHTF/SG1/N70:2011 includes the content of the label, shall i assue that GHTF/SG1/N70:201 fulfills the labeling requirement as per ISO 13485?
Looking forward for your response.
Best,
Rabel Talpur
Dear Rabel,
ISO 13485 does not specify the content of your labeling, that is correct. This something you will find in the national laws, regulations and ordinances.
The GHTF document is a good starting point for your labeling, but it does not necessarily cover all the national requirements – for the PMDA in Japan, you might want to consult the
– Act on Securing Quality, Efficacy and Safety of Products Including Pharmaceuticals and Medical Devices (https://www.japaneselawtranslation.go.jp/en/laws/view/3213#je_ch9sc1at1),
– Regulation for Enforcement of the Act on Securing Quality, Efficacy and Safety of Products Including Pharmaceuticals and Medical Devices (https://www.japaneselawtranslation.go.jp/en/laws/view/3215)
– Order for Enforcement of the Act on Securing Quality, Efficacy and Safety of Products Including Pharmaceuticals and Medical Devices (https://www.japaneselawtranslation.go.jp/en/laws/view/3214#je_ch7at4)
I hope this gives you a brief overview about the labeling process for the PMDA. Let us know if you require more specific support, we would be happy to help!
Kind regards
Christopher Seib
Vielen Dank erstmal für Ihre wertvollen Beiträge.
Wie sieht es bei der Priorisierung von Informationen auf Labels aus?
Ein Beispiel:
Aufgrund von Platzproblemen können nicht alle Informationen direkt auf dem Produktlabel (ein Reagenz) angebracht werden und müssen teilweise auf die Verpackung ausgelagert werden. Wäre in diesem Fall die UDI oder die CE-Kennzeichnung (oder sogar IVD-Symbol) direkt auf dem Produkt zu priorisieren?
Gibt es hierzu ganz allgemein Gewichtungen/regulatorische Hilfestellungen (Risikobasierter Ansatz etc.)?
Vielen Dank für Ihre Einschätzung!
Beste Grüße
Lieber Anton,
vielen Dank für die spannende Frage!
Die IVDR gibt Ihnen in Anhang VI, Teil C, Artikel 4.2 zumindest den Hinweis, dass die UDI in begründeten Ausnahmen auch auf die nächsthöhere Verpackungsebene ausgelagert werden darf. Die Priorisierung der weiteren Informationen finden Sie in Artikel 6.1.2 der EN ISO 18113-2. Demnach können „Inhaltsangabe, Gebrauch für die In-vitro-Diagnostik und die Lagerungs- und Handhabungsbedingungen abgekürzt oder ausgeklammert werden“ – also entsprechend auch hier wieder auf die nächsthöhere Verpackungsebene verschoben werden.
Die weiteren Informationen müssen Sie jedoch auf dem Primäretikett unterbringen.
Herzliche Grüße
Christopher Seib
Liebes Johner Team
Wir verkaufen ein Zubehör der Klasse Is (reflektierende Kugeln, selbst als Medizinprodukt bewertet) an Systemanbieter (Navigationskameraanbieter). Dieses Zubehör (der einzelne Artikel) ist mit einer bestimmten Artikelnummer zugelassen. Nun möchten aber die Systemanbieter jeweils für ihr System verschiedene Verpackungskonfigurationen. D.h. ein Kunde möchte 4 Beutel mit z.B. je 3 Artikel drin, der nächste möchte lieber 3 Beutel mit z.B. je 6 Artikel drin.
Bis anhin haben wir für die verschiedenen Konfigurationen eigene interne Artikelnummern vergeben (z.B. REF xyz1 für Verpackung mit 12 Artikeln und abc2 für Verpackung mit 18 Produkten). Somit ist es für uns einfacher kundenspezifisch die Produkte abzupacken und zu etikettieren. Bis anhin haben wir zusätzlich jeweils noch die Artikelnummer, mit welcher das Produkt zugelassen wurde, auf das Etikett angebracht.
Ist es laut MDR nötig diese Artikelnummer, welche die Zulassung hat, aufs Produkt anzubringen oder würde es auch reichen, wenn nur die Artikelnummern der Verpackungskonfigurationen aufgeführt sind? Intern sind die Stücklisten und die Rückverfolgbarkeit im ERP gewährleistet.
Oder müsste auf dem Karton die zugelassene Artikelnummer und zusätzlich die Anzahl der verpackten Produkte drauf?
Vielen Dank für Ihre Stellungnahme und Support.
Liebe Frau Ermel,
nur sofern jedes Produkt für sich die Konformität als eigenständiges Medizinprodukt erfüllt – das heißt unter anderem, jedes Produkt ist eigenständig vollständig gekennzeichnet und es liegen alle nötigen Informationen bei – dürften Sie den Karton als reine Transportverpackung deklarieren.
In Ihrem Fall vermute ich, dass die Begleitinformationen und die Kennzeichnung erst auf Ebene des Kartons geliefert werden. Demnach stellt dieser die Primärverpackung für mehrere Produkte dar und muss die Anzahl der enthaltenen Produkte aufzeigen und eine eigene Artikelnummer (bzw. UDI-DI) bekommen. Spätestens bei der Vergabe der UDI-DI gemäß Anhang VI, Teil B, werden Sie sich auch für die Eintragung Ihrer Produkte in EUDAMED festlegen müssen, welche konkrete Konfiguration vorliegt und dies auch auf dem Karton auch entsprechend angeben müssen.
Falls ich den Sachverhalt nicht korrekt verstanden habe, können wir uns auch gerne direkt zu Ihren Produkten austauschen.
Herzliche Grüße
Christopher Seib
Sehr geehrtes Johner Institut Team
Gerne wollte ich mich erkundigen welche Angaben zwingend auf eine Marketing-Broschüre eines Klass IIa Medizinproduktes gehören.
Muss das Herstellersymbol (schwarze Fabrik), CE-Zeichen mit Kennnummer usw. drauf? Welche Kriterien gelten für die interne Freigabe einer Marketing-Broschüre? Gibt es Richtlinien oder sogar Verordnungen dazu?
Mit grossem Interesse sehe ich Ihrem Feedback entgegen und verbleibe in der Zwischenzeit mit freundlichen Grüssen
D. Romeo
Sehr geehrter Herr Romeo,
hier liefert Ihnen das Heilmittelwerbegesetz (https://www.gesetze-im-internet.de/heilmwerbg/BJNR006049965.html) in Artikel 4 eine Auflistung der Informationen, die Sie zwingend auf allen Werbematerialien anbringen müssen. Zusätzlich sind Werbematerialien auch als Teil des Labelings zu verstehen und müssen somit vorab (für Klasse IIa oder höher) durch die Benannte Stelle geprüft werden; damit tragen sie weiterhin auch ein CE-Zeichen mit der Nummer der Benannten Stelle.
Die Freigabe ist Ihnen intern selbst überlassen, ich würde jedoch empfehlen, dass zumindest eine Prüfung durch einen Regulatory-Mitarbeiter erfolgt.
Herzliche Grüße
Christopher Seib
Sehr geehrtes Johner Team,
OEM PLM Konfiguration unter der MDR. Unsere Frage: Was muss auf der Etikette des Händlers an Daten des OEM enthalten sein?
PLM: Firmenname, Adresse
PLM : Artikel Nummer, eigene
PLM : LOT-Nummer, eigene
PLM: http://www.PLM.com, IFU, eigene
OEM: Hersteller Adresse und Firmensymbol, CE-Zeichen
OEM hat eine QSV-Vereinbarung mit dem PLM, OEM hat alle Produkte auf der EUDAMED hochgeladen!
Unsere Frage:
Die UDI-DI, wie muss diese generiert sein, mit den Daten des OEM oder des PLM? Wie erfolgt die Rückverfolgung des Produkts über die Eudamed, wenn der PLM nur seine eigene Artikelnummer und Lotnummer angeruckt hat.
Gibt es eine Anweisung, welche definiert, welche Daten des OEM und des PLM bei dieser Konstellation auf der Etikette erfasst werden müssen?
Vielen Dank für Ihre Stellungnahme zu diesem komplexen Gebiet.
Freundliche Grüsse,
HP.Hutter
EDENTA Etablissement, Schaanwald
Sehr geehrter Herr Hutter,
die MDR kennt die Konstellation des OEM PLM nicht (mehr) und verhindert eigentlich auch, dass eine solche Konfiguration überhaupt möglich ist (siehe auch https://www.johner-institut.de/blog/regulatory-affairs/oem-original-equipment-manufacturer/). Der OEM hat das Produkt in EUDAMED registriert und ist somit der Hersteller unter der MDR. Eine Veränderung des Labelings des Herstellers wäre nicht zulässig, der PLM kann höchstens ein weiteres Etikett mit eigenen Informationen anbringen, darf jedoch die ursprünglichen Informationen nicht verdecken. Damit ist auch klar, dass der OEM für die Registrierung eine UDI-DI mit den eigenen Informationen erstellen muss, ansonsten wäre eine Registrierung in EUDAMED nicht möglich und das Produkt dürfte kein CE-Zeichen tragen. Eine eigene IFU des PLM wäre ebenfalls nicht konform zur MDR.
Eine Alternative wäre, dass der PLM die Funktionen eines Händlers gemäß Artikel 14 der MDR übernimmt.
Herzliche Grüße
Christopher Seib
Sehr geehrtes Team des Johner Instituts,
wir sind Importeur und Händler für Medizinprodukte und verkaufen sowohl an Endkunden (z.B. Arztpraxen) als auch an andere Händler. Einer unserer Händler möchte, dass der Hersteller seinen Namen und Kontaktdaten auf die für ihn bestimmten Verpackungen aufdruckt, was wiederum wir aufgrund des direkten Kontaktes mit dem Hersteller in die Wege leiten sollen. Dies würde bedeuten, dass dann zusätzlich zum Hersteller, EC Rep und Importeur ein viertes Feld mit Daten des Händlers aufgedruckt wäre.
Wäre dies in Bezug auf die MDR ein erlaubtes und unproblematisches Vorgehen, oder würden wir dadurch zusätzliche Verpflichtungen bzw. Risiken eingehen, die über unsere bestehenden Pflichten als Importeur und Händler hinausgehen?
Wären bei diesem Vorgehen ggf. Unterscheidungen zwischen Produkt- und Umverpackungen notwendig?
Wie verhält es sich, wenn der Name, z.B. der des Händlers oder des Importeurs ein Logo (kein Markenname; nur ein Firmenlogo) enthält?
Vielen Dank für Ihre Antwort
Andrea Kiecker
Sehr geehrte Frau Kiecker,
prinzipiell sehe ich hier kein Problem, solange weiterhin klar ist, wer der Hersteller des Produktes ist und wer der Händler. Sie selbst müssen als Importeur ebenfalls ersichtlich sein. Gemäß Artikel 16 (2) b) würde ich dies nicht als Änderung des Produktes beurteilen, da keinerlei Risiken entstehen (solange keine der ursprünglichen Informationen verändert oder verdeckt werden). Die Verpackungsebene spielt dabei keine Rolle. Um klar zu machen, dass das entsprechende Unternehmen wirklich „nur“ Händler ist, würde ich empfehlen, dieses auch vollständig mit Adresse und dem zugehörigen Symbol aus der ISO 15223-1 auszuweisen und nicht nur über ein Logo, um eine mögliche Verwechslung direkt auszuschließen.
Herzliche Grüße
Christopher Seib
Sehr geehrter Herr Seib,
bei meiner Frage geht es um die Kennzeichnung am Produkt für Kompressionsstrümpfe.
Kompressionsstrümpfe werden idealerweise paarweise abgegeben. Die Produktkennzeichnung erfolgt mittels Einnähetikett.
Ist es dann MDR-konform ,nur EINEN Strumpf mit diesem zu versehen? Oder besteht die Konformität nur, wenn beide Strümpfe per Einnähetikett gekennzeichnet sind.
Bei uns gehen die Meinungen dazu auseinander.
M.E. ist es notwendig, BEIDE Strümpfe mittels Einnähetikett zu kennzeichnen, da jeder einzelne Strumpf als Medizinprodukt angesehen werden kann.
Ich bin auf Ihre Einschätzung gespannt.
Vielen Dank dafür im Voraus.
Mit freundlichen Grüßen
Christine Graß
Sehr geehrte Frau Graß,
hier kommt es vor allem darauf an, wie das Produkt bereitgestellt wird. Ich vermute, beide Strümpfe werden ausschließlich als Set verkauft. In diesem Fall spricht die MDR davon, das Produkt vollständig zu kennzeichnen – dies wäre über ein einziges Einnähetikett abgedeckt. Kann es bei sachgemäßer Anwendung zu einer Verwechslung kommen oder gehen Informationen verloren? Meiner Meinung nach nicht, da die Kompressionsstrümpfe immer als Paar angewendet werden. Sollte dem nicht so sein und jeder Strumpf als eigenes Medizinprodukt separat vertrieben bzw. angewendet werden, so wären auch beide getrennt zu kennzeichnen.
Stellen Sie wiederum ein Risiko fest (sicherheitsrelevante Informationen werden nicht zur Kenntnis genommen; möglicher einseitiger Einsatz / auch bei amputierten Patienten denkbar) so sollten Sie als entsprechende Maßnahme eine beidseitige Kennzeichnung in Betracht ziehen.
Ich hoffe sehr, das hilft Ihnen weiter!
Herzliche Grüße
Christopher Seib
Sehr geehrter Herr Seib,
vielen Dank für Ihre Antwort und Einschätzung. Sie haben mir damit weitergeholfen und haben meine Bedenken bzgl. einer Nichtkonformität ausgeräumt.
Danke dafür!
Viele Grüße
Christine Graß
Liebes Johner Team, lieber Herr Johner,
danke für die vielen hilfreichen Antworten, die Sie und Ihr Team bisher geschrieben haben.
Ich habe eine eher generelle Frage zu UDIs.
Wir haben ein Medizinprodukt, dass aus zwei gleich-aussehenden Komponenten (Komponente 1 (,,oben“) und Komponente 2 (,,unten“)) und einem Verbindungskabel besteht. Das zugelassene Produkt (bzw. die DoC) bezieht sich auf das Set und enthält eine Teileliste worin oben genannte Komponenten aufgelistet sind. Die Komponenten tragen jeweils ein Label mit UDI (DI & PI) bezogen auf die jeweilige Komponente auf der die Labels aufgebracht sind. Auf einem der Komponenten ist ein Zusätzliches Label aufgebracht, dass den Namen, die Revision und die Artikelnummer des zugelassenen Sets enthält; Dieses zusätzliche Set-Label hat jedoch keine SN, kein Produktionsdatum und keinen UDI.
Ist dieses UDI-,,Setup“ vereinbar mit MDR?
Liebe Grüße,
Sebastian S.
Lieber Herr S.,
ich vermute, beide Komponenten sind kommerziell nicht separat erhältlich? In dem Fall müssten diese eigentlich keine eigene UDI tragen, das Set als Ganzes jedoch schon. Diese könnten Sie dann auf einer der beiden Komponenten anbringen. Sind beide Komponenten einzeln erhältlich, muss dennoch auch eine weitere UDI auf „Set-Ebene“ vergeben werden, da dieses ein eigenes Medizinprodukt darstellt.
Eine Ferndiagnose ist hier meist etwas schwierig – melden Sie sich gerne, wenn wir uns das einmal konkret zusammen anschauen sollen.
Herzliche Grüße
Christopher Seib
Liebes Johner- Team!
Wir planen, die Abgabe von Mustern (Klasse III Medizinprodukt) an unsere Sales Reps bzw. das „Schicksal“ dieser Muster besser zu nachzuverfolgen und würden daher in Zukunft die Muster (die Produkte verfügen über CE nach MDD und bald auch MDR) mit einem Sticker „Doctors Sample – not for sale“ versehen. Muss diese Änderung Ihrer Ansicht nach vorab dem NB vorgelegt werden bzw. handelt es sich dabei um eine zusätzliche Produktvariante? Das Zusatzlabel, mit dem die Produkte auf der Verpackung gekennzeichnet werden sollen, wäre so gestaltet das keine relevanten Informationen der Verpackung verdeckt werden.
Herzlichen Dank!
Beste Grüße
Stefan Scheider
Lieber Herr Scheider,
ein Muster stellt an sich kein Medizinprodukt dar, da es nicht dazu gedacht ist, einen medizinischen Zweck zu erfüllen. Entsprechend wären weder die MDR einzuhalten noch eine Benannte Stelle zu informieren.
Wichtig ist jedoch, dass auch wirklich nichts auf ein Medizinprodukt hindeutet. So darf kein CE-Zeichen (mehr) zu erkennen sein, und auch das MD-Symbol wäre irreführend. Beachten Sie auch, dass die Gebrauchsanweisung keine Leistungsdaten mehr enthält, sondern maximal Informationen wie die Installation oder ähnliches. Ich würde statt „not for sale“ noch einen Schritt weiter gehen und „not for diagnostic / therapeutic use“ schreiben.
Herzliche Grüße
Christopher Seib
Liebes Johner-Team,
eine kurze Frage. Ist es möglich ein Typenschild mit allgemeinen Warnsymbolen die nicht spezifisch sind (wie Datum und Seriennummer) auf die Batteriefachabdeckung zu kleben. Dieses Fach wäre allerdings abnehmbar und könnte somit verloren gehen etc. Konkret geht es um ein Platzproblem bei einem kleinen Medizinproduktgehäuse.
Vielen Dank.
Guten Morgen,
die Batteriefachabdeckung stellt eine Komponente Ihres Medizinproduktes dar, welche bei bestimmungsgemäßem Gebrauch über die gesamte Lebensdauer hinweg erhalten bleiben sollte, allein schon, um Anwenderrisiken zu minimieren. Sie als Hersteller müssen dies im Rahmen der Validierung (zum Beispiel nach mehrmaligem Öffnen und Schließen) nachweisen können. Dies wäre zwar eventuell nicht der präferierte Ort zum Anbringen der Informationen (siehe auch ISO 20417, Anhang B), doch bei Platzmangel sehe ich hier kein regulatorisches Hindernis.
Herzliche Grüße
Christopher Seib
Guten Tag zusammen,
ich habe eine Frage an das Johner Team.
Müssen auf sterilen Peelpackungen die schriftliche Bezeichnung =>
Nicht verwenden, wenn die Verpackung beschädigt oder geöffnet ist.
Dass das Symbol nach DIN EN ISO 15223-1 oder ISO 7000-2606
auf die sterile Peelpackungen vorhanden sein muss ist für mich verständlich.
Aber wie sieht es mit den Textblock aus? Es gibt diverse Hersteller die den
Text «Nicht verwenden, wenn die Verpackung beschädigt ist» was sagt hier die MDR?
Vielen Dank im Voraus für Ihre Unterstützung
Mit freundlichen Grüßen
A.Brandt
Guten Morgen Herr Brandt,
um genau diese Textblöcke zu vermeiden gibt es die harmonisierte EN ISO 15223-1! Eine Beschreibung des Symbols auf dem Etikett ist damit nicht mehr nötig, sollte jedoch in der Gebrauchsanweisung erfolgen. Die MDR selbst macht dazu nur wenig konkrete Vorgaben, außer dass ein entsprechender Hinweis erfolgen muss, und zwar in Form von international anerkannten Symbolen, sofern es welche gibt. Vielmehr sind es die nationalen Regularien, die eine Übersetzung des Textes in eine anerkannte Sprache fordern.
Herzliche Grüße
Christopher Seib
Hallo Herr Seib,
vielen Dank für Antort, diese ist für mich sehr hilfreich gewesen. Vieleicht könnten Sie mir noch eine Frage zum Begriff „Labelling“ beantworten. Ich habe Layouts von sterilen Copolymer Handschuhen gesehen, wo die CE-Kennzeichnung mit benannter Stelle auf der Peelpackung und der Box vorhanden ist. Auf dem Transportkarton ist nur die CE-Kennzeichnung ohne benannte Stelle. Gibt es eine Richtlinie oder Verordnung, die besagt das auf allen Verpackungseinheiten die CE-Kennzeichnung mit benannter Stelle sein muss? Oder reicht Peelpackung und Box aus?
Vielen Dank im Voraus für Ihre Unterstützung
Herzliche Grüße
A.Brandt
Hallo Herr Brandt,
sehr gerne, das freut mich!
Artikel 20(3) der MDR beschreibt: „Die CE-Kennzeichnung erscheint auch in jeder Gebrauchsanweisung und auf jeder Handelsverpackung.“ Diese muss dann auch einheitlich (sprich immer mit bzw. ohne Nummer der Benannten Stelle) angebracht werden. Handelt es sich um einen reinen Transportcontainer, so ist dieser nicht als Teil des Produktes zu bewerten und darf entsprechend keine CE-Kennzeichnung tragen. Andernfalls muss analog zum Produkt das CE mit Nummer vorliegen.
Herzliche Grüße
Christopher Seib
Hallo Herr Seib,
herzlichen Dank für Ihre Antwort. Die Informationen waren sehr wieder hilfreich.
Schöne Grüße
A.Brandt
Sehr geehrter Herr Seib,
Es geht um die Etikettierung eines Is Medizinprodukt Zubehörs. Die MDR schreibt vor, dass Zubehör als eigenständiges Medizinprodukt behandelt werden soll.
In Anhang I Aritkel 23.2 q) steht „einen Hinweis, dass es sich bei dem Produkt um ein Medizinprodukt handelt. Ist das Produkt lediglich für klinische Prüfungen vorgesehen, die Aufschrift „ausschließlich für klinische Prüfungen“
Muss Zubehör ebenfalls als Medizinprodukt auf dem Etikett der Verpackung (z.B durchs MD symbol) gekennzeichnet werden? Das Zubehör wird separat vom Medizinprodukt vertrieben.
Vielen Dank im Voraus
Mit freundlichen Grüßen,
L. Guthmann
Sehr geehrter Herr Guthmann,
machen Sie am besten keine so Trennung zwischen Zubehör und Medizinprodukt. Regulatorisch gesehen ist jedes Zubehör ein eigenes Medizinprodukt und muss entsprechend alle Anforderungen identisch erfüllen. Daher trägt auch das Zubehör das CE-Zeichen und ein MD-Symbol.
Herzliche Grüße
Christopher Seib
Liebes Johner-Team,
die Verpackung unseres Produkts besteht aus einer Tube und einer Faltschachtel. Ist es bzgl. der Sprachenregelung möglich, dass die Tube Angaben auf deutsch enthält und die Faltschachtel und die Gebrauchsinformation die notwendigen Angaben in der jeweiligen Landessprache?
Vielen Dank und viele Grüße
Liebe Lisa,
das ist leider nicht ganz einfach zu beantworten. Generell sollten sich auf der Tube (direkt auf dem Produkt) alle von der MDR geforderten Informationen finden. Teile Können bei Platzmangel auf die Verpackung ausgelagert werden. Die nationalen Vorgaben fordern nun jedoch, das alle Informationen, die die MDR fordert, in Landessprache vorliegen müssen, also wäre eine Deutsche Direkt-Kennzeichnung nicht ausreichend, auch wenn die Sekundärverpackung mehrsprachig vorliegt. Vor allem, da das Produkt höchstwahrscheinlich an einen Laien geht.
Wäre es hier nicht möglich, zumindest direkt auf der Tube nur mit Symbolen nach der EN ISO 15223-1 zu arbeiten?
Herzliche Grüße
Christopher Seib
Guten Tag zusammen,
ich habe eine Frage an das Johner Team.
Wir sind OEM Hersteller für Medizintechnik und verkaufen unsere Produkte zugelassen an Distributoren.
Ist es nun Pflicht das der Distributor mit Name und Adresse auf dem Label/Etikett gelistet ist, oder reicht es wenn wir als Hersteller und ggf. der EC Rep darauf stehen?
Danke im Voraus und freundliche Grüsse
G. Mäder
Guten Morgen Herr Mäder,
solange Ihr Distributor keine Tätigkeiten nach Artikel 16 übernimmt muss dieser auf dem Etikett nicht genannt werden. Das ist nur für den Importeur bzw. den EU-Repräsentanten verpflichtend.
Herzliche Grüße
Christopher Seib
Hallo zusammen,
ich habe eine Frage an das Johner Team. Ich möchte gerne wissen, ob der Hersteller auf Etiketten „Brand name“ definieren soll.
Danke im Voraus und freundliche Grüsse
Aslani
Hallo Aslani,
die MDR spricht von Name oder Handelsname auf dem Etikett. Prinzipiell ist beides möglich, sofern der Anwender darüber identifizieren kann, welches Produkte gerade vorliegt. Entscheidend ist, dass der Handelsname spezifisch für das Produkt ist (siehe auch Kapitel 5.6 der ISO 20417). Außerdem ist wichtig zu beachten, dass dem Anwender über den Handelsnamen (im Gegensatz zu den meisten Produktnamen) eventuell nicht klar ist, worum es sich bei dem Produkt handelt, daher könnte eine zusätzliche Erklärung nötig werden.
Generell würde ich mir hierfür immer die Frage stellen: Was genau hat der Anwender bei uns bestellt? Läuft das wirklich über den Handelsnamen? Wenn ja, dann sollte sich dieser auch auf dem Produktetikett wiederfinden, ansonsten reicht auch der Produktname.
Herzliche Grüße
Christopher Seib
Sehr geehrtes Johner-Team,
die Etiketten von Medizinprodukten der Klasse I und IIa, die weltweit vertrieben werden, werden verändert.
Neue Symbole, UDI werden hinzugefügt.
Was muss beachtet werden (Informationsausmaß an Händler, Risikomanagement) ?
Vielen Dank im Voraus
Sehr geehrter Herr Mühe,
eine Änderung am Labeling ist eine Änderung am Produkt. D.h. Sie müssen die Konformität neu bewerten und bestätigen.
Die Bewertung wird v.a. sicherstellen müssen,
Beste Grüße, Christian Johner
Sehr geehrter Her Seib,
bitte gestatten Sie mir eine kurze Rückfrage zu Ihrer Antwort vom 18.04.2024: wäre die Vorgehensweise bzgl. „Doctors Sample“ in USA die gleiche oder müssten hier weitere Maßnahmen getroffen werden?
Beste Grüße
Stefan Scheider
Sehr geehrter Herr Schneider,
in der Theorie ja, in der Praxis könnte Ihnen jedoch der Zoll direkt einen Strich durch die Rechnung machen und das Produkt nicht ins Land lassen. Eventuell macht es Sinn, den Sachverhalt vorab mit dem Zoll zu klären.
Herzliche Grüße
Christopher Seib
Sehr geehrtes Team des Johner Instituts,
wir haben eine Frage zum Handling von Mustern/Proben für stoffliche Medizinprodukte.
Für Fertigarzneimittel ist in §47 (3) im AMG die Musterabgabe bzgl. Fertigarzneimitteln geregelt. Für Medizinprodukte gibt es keine Regelung – dürfen Muster (hier geht es explizit um stoffliche Medizinprodukte) abgegeben werden und wenn ja an wen? Auch an Patienten (was für Fertigarzneimittel und OTC-Präparate nicht möglich ist)? Müssen Muster der stofflichen Medizinprodukte der Benannten Stelle gemeldet werden?
Sehr geehrte Frau Killian,
die Abgabe von Mustern, die einen tatsächlichen medizinischen Zweck erfüllen (sprich unter den Geltungsbereich der MDR fallen und dazu gedacht sind, angewendet zu werden), fällt unter den Begriff der Bereitstellung (siehe Kapitel 2.2 Amtsblatt C 247 der Europäischen Union). Damit muss das Produkt – egal ob stofflich oder nicht – zum Zeitpunkt der Abgabe die Anforderungen der MDR erfüllen. Hier spielt der Anwender keine Rolle.
Die Abgabe von Mustern, welche nicht dafür gedacht sind, einen medizinischen Zweck zu erfüllen, würden unter das Heilmittelwerbegesetz , Artikel 11.14 fallen und sind nur für Angehörige der Gesundheitsberufe zulässig.
Herzliche Grüße
Christopher Seib
Guten Tag,
ich habe eine Frage zur nachgelagerten, praktischen Verwendung der (korrekten und vollständigen) Zweckbestimmung als Teil der „vom Hersteller bereitzustellenden Informationen“ gemäß MDR.
Die MDR beschreibt ja die Zweckbestimmung wie folgt:
…
„Zweckbestimmung“ bezeichnet die Verwendung, für die ein Produkt entsprechend den Angaben des Herstellers auf der Kennzeichnung, in der Gebrauchsanweisung oder dem Werbe- oder Verkaufsmaterial bzw. den Werbe- oder Verkaufsangaben und seinen Angaben bei der klinischen Bewertung bestimmt ist;
…
Dies erweckt den Eindruck, dass auf der Kennzeichnung (z.B. Etiketten) stets die Zweckbestimmung in Gänze zu nennen wäre.
Ich interpretiere es eher so, dass die Angaben auf der Produktkennzeichnung und in den Werbematerialien der Zweckbestimmung zumindest in keiner Weise widersprechen dürfen.
Weiter steht in der MDR, 23.2 „Angaben auf der Kennzeichnung“ jedoch:
Die Kennzeichnung enthält alle folgenden Angaben:
…
alle unbedingt erforderlichen Angaben, aus denen der Anwender ersehen kann, worum es sich bei dem Produkt, dem Packungsinhalt sowie der Zweckbestimmung eines Produkts, sofern diese für den Anwender nicht offensichtlich ist, handelt;
…
In der Praxis scheint es dennoch aus nachvollziehbaren Gründen kaum Hersteller zu geben, welche die komplette Zweckbestimmung tatsächlich auf ein MDR-Produktetikett bringen.
Ist davon auszugehen, dass ein (exakt) beschreibender Produktname auf dem Etikett, z.B. „Spritzenpumpe“ in Verbindung mit dem Symbol „Gebrauchsanweisung beachten“, in welcher die Zweckbestimmung sowie die Anwendergruppe enthalten ist, in jedem Fall die regulatorischen Anforderungen der MDR erfüllt?
Mit freundlichen Grüßen
A. Perez Pulgar
Guten Morgen,
die Bezeichnung „Intended purpose“ ist an dieser Stelle tatsächlich nicht ganz glücklich. Im Endeffekt ist es wichtig, dass Ihr Anwender weiß, wozu dieses Produkt gedacht ist – die vollständige Erklärung erfolgt dann, wie Sie bereits erwähnt haben, in der Gebrauchsanweisung.
Das Etikett dient in erster Linie der sicheren Identifikation des Produktes, und das wäre über die Bezeichnung „Spritzenpumpe“ gegeben. Damit wäre für den Anwender nun auch (die grobe) Zweckbestimmung offensichtlich – zumindest hoffe ich das bei einem professionellen Anwender. Bei einem Laien würde ich die Zweckbestimmung jedoch deutlich ausführlicher aufführen (zum Beispiel: Corona-Antigen Test für den Selbsttest zur Detektion von…).
In der Praxis wird hier üblicherweise die Produktbezeichnung verwendet, um diese Anforderung zu erfüllen und dies wird so auch akzeptiert. Verwechseln Sie jedoch nicht den Intended Purpose mit dem Intended Use (https://www.johner-institut.de/blog/regulatory-affairs/zweckbestimmung/).
Herzliche Grüße
Christopher Seib
Guten Tag, ich habe folgende Frage zur Kennzeichnung: Darf ich das Etikett eines Medizinproduktes entfernen, wenn ich das Produkt als Komponente meines eigenen Medizinproduktes verwende?
Szenario: Ich habe ein Medizinprodukt der Klasse I mit dem Produktnamen „Produkt 1“, das aus verschiedenen Komponenten besteht und ich führe die Konformitätserklärung mit allen diesen Komponenten durch und stelle die Konformität fest. Das Medizinprodukt als Einheit erhält ein Etikett mit dem CE-Zeichen.
Wenn nun eine der oben genannten Komponenten meines Medizinproduktes selbst ein Medizinprodukt ist, darf ich dann das ursprüngliche Etikett von dieser Komponente entfernen, da diese ein Teil meines Medizinproduktes ist? Darf ich mein Etikett darauf anbringen?
Ich wäre Ihnen sehr dankbar, wenn Sie mir in dieser Angelegenheit helfen könnten.
Mit freundlichen Grüßen
E. Jaberi
Guten Tag!
Indem Sie das Medizinprodukt als Komponente in Ihr eigenes Produkt integrieren, ändern Sie (höchstwahrscheinlich) auch die ursprüngliche Zweckbestimmung. Sie selbst übernehmen komplett eigenständig die Verantwortung, dass diese Komponente nun korrekt funktioniert und müssen – wie Sie bereits richtig beschrieben haben – eine eigene Konformitätsbewertung durchführen. In diesem Fall ist es sogar nötig, dass Sie das originale Etikett entfernen, da die dort enthaltenen Informationen nun nicht mehr korrekt sind. Die Komponente stellt durch das Einbauen in Ihr Produkt kein eigenständiges Medizinprodukt mehr dar.
Herzliche Grüße
Christopher Seib