
Medizinprodukte über das Ende der Lebensdauer hinaus zu betreiben kann gefährlich und rechtlich problematisch sein. Daher sollten Hersteller die genaue Lebensdauer bei jedem Produkt präzise bestimmen. Doch könnten Sie auf die Schnelle sagen, wie Lebensdauer definiert oder nach welchen Kriterien sie bestimmt wird?
Verwandte Begriffe wie „Nutzungsdauer“, “Haltbarkeit”, “Betriebszeit” bzw. „Service Life“ und „Shelf Life“ stiften oft zusätzliche Verwirrung – genauso wie die Tatsache, dass Lebensdauer gar nicht zwangsläufig nur eine “Dauer” sein muss, sondern auch andere Faktoren eine Rolle spielen können.
Vermeiden Sie Ärger mit Behörden und Kosten oder sogar Schäden von Patienten und erfahren Sie in diesem Beitrag:
- warum es wichtig ist, die Lebensdauer zu bestimmen
- wie Sie die Lebensdauer definieren und festlegen können
- wie Lebensdauer von anderen Begriffen wie Service-Zeit oder Betriebsdauer abzugrenzen ist und
- welche (regulatorischen) Anforderungen Sie als Hersteller und Betreiber erfüllen müssen.
1. Grundlegendes zur Lebensdauer
1.1 Begrifflichkeiten: Lebensdauer, Nutzungsdauer und mehr
Die Lebensdauer von Medizinprodukten ist der Zeitraum, in dem ein Produkt sicher und leistungsfähig ist. Naturgemäß ändern sich Produkteigenschaften mit der Zeit, etwa durch Alterung oder Abnutzung. Sind Sicherheit und Leistungsfähigkeit dadurch nicht mehr gegeben, können große Probleme entstehen, etwa:
- Eine Dialysemaschine wird über die Zeit in der Flüssigkeitsbilanzierung ungenau, weil Dichtungsmaterialien altern und Flüssigkeitsverluste auftreten.
- Kunststoffe werden spröde und verlieren ihre Bruchstabilität.
- Elektronische Bauteile können nach vielen Schaltvorgängen ausfallen.
Die Lebensdauer von Medizinprodukten ist sowohl für Hersteller als auch für Betreiber relevant.
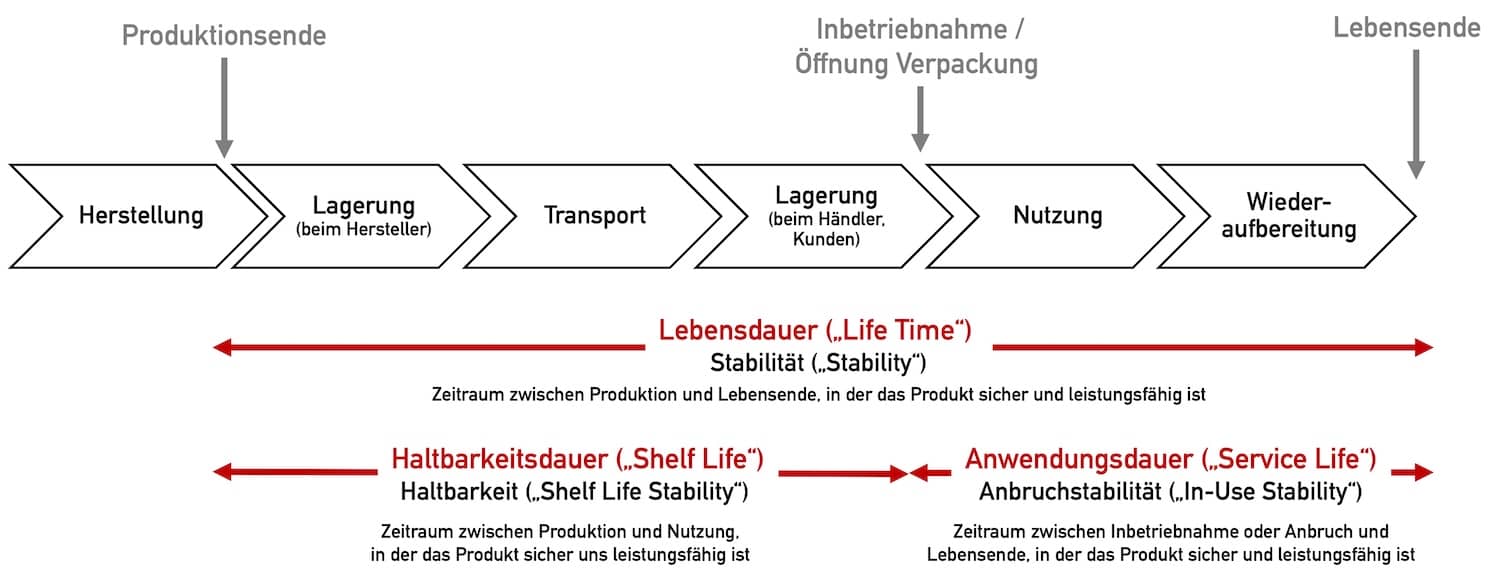
Die Begrifflichkeiten werden oft nicht präzise verwendet und Dauer (Zeiträume) und Stabilitätskriterien nicht unterschieden. Abbildung 1 markiert die Dauern (Zeiträume) in rot, die Arten der Stabilität in schwarz.
Die weiteren Begrifflichkeiten wie Nutzungsdauer und Haltbarkeitsdauer definiert und erklärt der Abschnitt 3.
Die Nutzungsdauer entspricht in Abbildung 1 der „Anwendungsdauer“.
1.2 Relevanz
1.2.1 Relevanz für Hersteller
Hersteller müssen die Lebensdauer als Teil der formulierten Zweckbestimmung kennen.
- Sie benötigen die Lebensdauer für das Risikomanagement (Anhang I MDR; ISO 14971). Sie müssen dort analysieren, ob das Produkt über die gesamte Lebensdauer sicher und leistungsfähig ist.
- Die Lebensdauer korreliert mit der Gesamtzahl an Anwendungen. Beide bestimmen die Risikoakzeptanzmatrix. Die Klasse der unwahrscheinlichsten Schäden (meist die unterste Zeile) sollte Wahrscheinlichkeiten entsprechen, die während aller Anwendungen über die komplette Lebensdauer nicht zu erwarten sind.
- Die vorgesehene Lebensdauer entscheidet bereits bei der Entwicklung über die Auswahl von Komponenten, Materialien und Technologien
- Die Lebensdauer und nötige Service-Intervalle sind eng miteinander verknüpft. Diese Service-Intervalle dokumentiert der Hersteller in den Begleitmaterialien.
1.2.2 Relevanz für Betreiber
Läuft die angegebene Lebensdauer des Produktes ab, müssen auch Betreiber reagieren (z. B. durch Außerbetriebnahme). Damit tun sich die Betreiber häufig schwer, denn die Produkte funktionieren augenscheinlich tadellos. Sie können nicht erkennen, dass die Sicherheit oder Leistungsfähigkeit beeinträchtigt ist. Das dürfte auch der Grund sein, warum viele Betreiber mit der Außerbetriebnahme hadern.
Noch schwieriger ist der Fall, wenn der Hersteller die Lebensdauer nicht in den Begleitpapieren angegeben hat. Woher soll der Betreiber in diesem Fall wissen, ab wann die Sicherheit und Leistungsfähigkeit nicht mehr gegeben sind?
Selbst wenn Betreiber der Meinung sind, ihr Produkt funktioniere noch tadellos: Entscheidend ist die vom Hersteller angegebenen Lebensdauer.
1.2.3 Relevanz für Kunden
Auch für Kunden ist die Lebensdauer eines Produktes ein wichtiges Kriterium. Sie ist nämlich in vielen Fällen ein Erwägungsgrund bei der Kaufentscheidung: Hat ein Einkäufer eines Krankenhauses die Wahl zwischen zwei sonst vergleichbaren Produkten, wird er sich für das Produkt mit der längeren Lebensdauer entscheiden.
2. Definition von Lebensdauer
So wichtig es für Hersteller, Betreiber, Kunden und Behörden ist, zu wissen, was die Lebensdauer eines Medizinproduktes ist, so schwierig ist es, genau zu definieren (und damit festzulegen), was “Lebensdauer” eigentlich ausmacht. Rechtliche Regelungen bleiben meist vage, und viele Begriffe werden synonym zu “Lebensdauer” verwendet.
2.1 MDR
Die EU-Verordnung 2017/745 (MDR) definiert Lebensdauer nicht explizit. Allerdings fasst sie in Anhang I grundlegende Sicherheits- und Leistungsanforderungen zusammen und definiert dabei ganz nebenbei und indirekt den Begriff „Lebensdauer“:
Die Merkmale und die Leistung des Produkts dürfen nicht soweit beeinträchtigt werden, dass die Gesundheit oder die Sicherheit des Patienten oder Anwenders oder gegebenenfalls Dritter während der Lebensdauer des Produkts gefährdet wird, wenn das Produkt Belastungen ausgesetzt wird, wie sie unter normalen Verwendungsbedingungen auftreten können, und es ordnungsgemäß entsprechend den Anweisungen des Herstellers instand gehalten wurde.
Quelle: MDR Anhang I Kap. I Nr. 6
„Entsprechend den Anweisungen des Herstellers“ meint dabei beispielsweise Wartungsanforderungen mit Prüfungen durch den Anwender, den präventiven Austausch von Komponenten mit geringerer Lebensdauer oder Kalibrierungen.
Interessant ist dabei, dass der englische Originaltext inhaltlich abweicht:
The characteristics and performance of a device shall not be adversely affected to such a degree that the health or safety of the patient or the user and, where applicable, of other persons are compromised during the lifetime of the device, as indicated by the manufacturer, when the device is subjected to the stresses which can occur during normal conditions of use and has been properly maintained in accordance with the manufacturer’s instructions.
Quelle: MDR Anhang I Kap. I Nr. 6
“As indicated by the manufacturer” bezieht sich auf “the lifetime of the device” und man könnte damit die Forderung zur Angabe der Lebensdauer durch den Hersteller verstehen.
2.2 IMDRF
Während die MDR den Begriff Lebensdauer nicht explizit definiert, findet sich im Dokument „Essential Principles of Safety and Performance“ des International Medical Device Regulators Forum (IMDRF) eine genauere Erklärung. Demnach ist Lebensdauer
“der vom Hersteller angegebene Zeitraum, in dem das Medizinprodukt oder IVD-Medizinprodukt voraussichtlich sicher und wirksam verwendet werden kann. (Time period specified by the manufacturer during which the medical device or IVD medical device is expected to maintain safe and effective use.)”
Quelle: IMDRF Essential Principles of Safety and Performance
Das alte IMDRF-Dokument „GHTF SG2 N21 R8 May 1999“ macht in Kapitel 2.3 die Lebensdauer dagegen auch von der vorgesehenen Nutzungsdauer abhängig: “die Zeit oder Nutzung, die ein Produkt funktionsfähig bleiben soll, nachdem es hergestellt, in Betrieb genommen und wie angegeben gewartet wurde.”
Service life is defined as: the time or usage that a device is intended to remain functional after it is manufactured, placed into use, and maintained as specified.
Quelle: GHTF SG2 N21 R8 May 1999 Kap. 2.3
2.3 IEC 60601-1 (zu aktiven Medizinprodukten)
Für die aktiven Medizinprodukte definiert die IEC 60601-1 die “zu erwartende Betriebs-Lebensdauer” als
vom Hersteller definierter Zeitraum, während dem zu erwarten ist, dass das ME-Gerät oder ME-System für den Gebrauch sicher bleibt (d. h., dass es die Basissicherheit und die wesentlichen Leistungsmerkmale aufrechterhält)
Quelle: IEC 60601-1
2.4 Wie Sie mit den verschiedenen Definitionen umgehen sollten
Da der Nachweis für die Erfüllung der grundlegenden Sicherheits- und Leistungsanforderung der MDR nach Anhang I Abschnitt 6 in der Technischen Dokumentation des Produktes erbracht werden muss, empfehlen wir, dort auch eine Interpretation des Begriffes “Lebensdauer” zu dokumentieren.
Ein Hersteller könnte die Lebensdauer etwa als Anzahl der Anwendungen (“300 Anwendungen”), ein anderer als Zeitspanne (“bis 5 Jahre nach der Inbetriebnahme”) oder bis zu einem bestimmten Zeitpunkt (“bis 12/2025”) verstehen. Auch Kombinationen sind möglich: 5 Jahre oder 1000 Behandlungen, je nachdem, welches Kriterium zuerst eintritt.
Wichtig ist, dass die Lebensdauer grundsätzlich nicht das Ergebnis der Produktentwicklung sein sollte. Vielmehr sollte diese vor Produktentwicklung festgelegt werden und dort als Vorgabe (“Design Input”) dienen.
Hiervon gibt es allerdings auch Ausnahmen: Etwa bei Reagenzien, bei denen Hersteller unter Umständen die Lebensdauer erst nach der Verifikation und Validation final festlegen können.
3. Weitere Begriffe
Wichtig ist auch zu wissen, welche Begriffe zwar oft im Zusammenhang mit Lebensdauer verwendet werden, aber dennoch nicht damit verwechselt werden sollten.
3.1 Mindesthaltbarkeit
Die Angabe der “Mindesthaltbarkeit” kennen die meisten aus dem Lebensmittelbereich. Im Umfeld von Medizinprodukten wird dieser Begriff nicht (mehr) verwendet. Im Gegensatz zu Lebensmitteln, bei denen das Mindesthaltbarkeitsdatum eher einen Orientierungscharakter hat, sind Haltbarkeitsdauer und Lebensdauer bei Medizinprodukten verbindlich.
Das heißt, dass nach Ablauf des angegebenen Zeitraums bzw. bei Erreichen eines angegebenen Haltbarkeitsdatums (siehe ISO 15223: “use-by-date”) Sicherheit und/oder Leistungsfähigkeit nicht mehr gegeben sind und das Produkt deshalb nicht mehr verwendet werden darf.
3.2 Stabilität, einschließlich Haltbarkeitsdauer („stability, including shelf life“)
Die Haltbarkeitsdauer („shelf life”) wird von der ZLG definiert.
Vom Hersteller bei Einhaltung der spezifizierten Lagerungs- und Transportbedingungen angegebene Haltbarkeit bis zur ersten Verwendung des Produktes
ZLG Dokument (3.3 A 5, Haltbarkeit – Bewertung des festgelegten Shelf Life)
Mit der ersten Verwendung eines Produktes endet die Shelf Life, beispielsweise beim Öffnen einer Verpackung (z. B. dentale Füllungsmaterialen zur mehrfachen Portionierung bei mehreren Anwendungen).
Eine besondere Bedeutung hat die Haltbarkeitsdauer („shelf life“) bei Sterilprodukten, die während Transport und Lagerung bis zur ersten Verwendung steril bleiben müssen. Hier gibt der Hersteller an, bis zu welchem Zeitpunkt die Sterilität unter angegebenen Bedingungen erhalten bleibt.
Den Begriff “Stabilität” verwendet man im Allgemeinen, um eine Produkteigenschaft auszudrücken. Der Hersteller muss sowohl für die Haltbarkeitsdauer, als auch für die Lebensdauer die Stabilität (= Haltbarkeit) nachweisen (vgl. Abb. 1).
Da auch nach der ersten Verwendung das Medizinprodukt Beeinträchtigungen der Sicherheit und Leistungsfähigkeit erfahren kann, spricht man ab diesem Zeitpunkt bis zum Ende der Lebensdauer von Anbruchstabilität (“in-use stability”) (z. B. bei dentalen Füllungsmaterialen).
Die Lebensdauer des Produktes beginnt ab Produktionszeitpunkt und schließt deshalb die Haltbarkeitsdauer („shelf life“) mit ein.
3.3 Service-Zeit („end of service”)
Hier kommt es regelmäßig zu Irritationen, da der Begriff „Service-Zeit“ als unglückliche deutsche Übersetzung der „Service Lifetime“ (= Lebensdauer) verwendet wird.
Service-Zeit ist jedoch nicht dasselbe wie Lebensdauer.
Zeitraum für den der Hersteller die Lieferbarkeit von Ersatzteilen und die Nachrüstbarkeit der Geräte zusagt.
Dieser Zeitraum wird vom Hersteller bzw. der Service-Organisation festgelegt und im regulatorischen Sinne nicht definiert. Es sei denn, der Hersteller kündigt Serviceleistungen vor Ablauf der Lebensdauer des Produktes. Dann kann das Medizinprodukt wegen fehlender Wartungs- und Reparaturarbeiten nicht mehr sicher und leistungsfähig betrieben werden und müsste unter Umständen vom Betreiber außer Betrieb gesetzt bzw. vom Markt zurückgerufen werden.
3.4 Betriebszeit
Als solche bezeichnet man die reine Dauer des aktiven Betriebs, also der tatsächlichen Nutzung eines Produktes. Diese ist nicht dasselbe wie die Lebensdauer, denn Lebensdauer hängt nicht zwingend von der tatsächlichen Nutzung ab.
Eine Dialysemaschine hat eine Lebensdauer von zehn Jahren. Selbst wenn das Gerät innerhalb dieser zehn Jahre nur für eine halbe Stunde genutzt wurde, ist die Lebensdauer am Ende der zehn Jahren abgelaufen.
Die Betriebszeit hängt also weniger vom Produkt ab, als vom Einsatzbereich und seinen Anwendern. Sie korreliert gewissermaßen mit der Lebensdauer, muss es aber nicht, weil auch Stillstandszeiten, Lagerung und Transport eine Alterung oder andere Effekte bedingen.
Weil die alterungsbedingte und die anwendungsbedingte Lebensdauer unterschiedlich sein können, gibt man – wie oben bereits erwähnt – manchmal beides an: 5 Jahre oder 1.000 Behandlungen, je nachdem, welches Kriterium zuerst eintritt.
3.5 Lebensende („end of life“)
Die IMDRF definiert das Lebensende im Kontext der Cybersecurity.
End of Life (EOL): Life cycle stage of a product starting when the manufacturer no longer sells the product beyond their useful life as defined by the manufacturer and the product has gone through a formal EOL process including notification to users.
Quelle: Cybersecurity des IMDRF
Es geht dabei also um den Zeitpunkt, zu dem ein Hersteller die Vermarktung eines Produktes einstellt. „Lebensende“ bezieht sich damit auf eine Produktlinie, nicht, wie bei der Lebensdauer, auf ein Einzelprodukt!
Die US-amerikanische FDA verwendet den Begriff “end of life (EOL)” allerdings anders.
Expected life of a device means the time that a device is expected to remain functional after it is placed into use. Certain implanted devices have specified „end of life“ (EOL) dates. Other devices are not labeled as to their respective EOL, but are expected to remain operational through activities such as maintenance, repairs, or upgrades, for an estimated period of time.
21 CFR 803.3 Buchstabe f
Damit sind im US-amerikanischen Raum „end of life“ und „Lebensdauer“ äquivalent.
3.6 Nutzungsdauer
Unter der Nutzungsdauer versteht man meist die Dauer, in der das Produkt nach dessen Inbetriebsnahme bzw. Öffnung der Verpackung bis zu dessen Lebensende genutzt werden. kann. Die Nutzungsdauer ist damit ein Synonym für die Anwendungsdauer bzw. „Service Life“ (s. Abbildung 1).
Gelegentlich wird die Nutzungsdauer auch als die Dauer verstanden, in der das Produkt nach Inbetriebnahme bzw. Öffnung der Verpackung genutzt werden kann, bevor eine Wiederaufbereitung notwendig wird. Das entspricht in Abbildung 1 der Länge des Pfeils „Nutzung“.
3.7 Zwischenfazit und -zusammenfassung
3.7.1 Begriffe
- Lebensdauer (“Service Life”): Gesamte Zeitspanne von Produktion bis Lebensende des Produktes, in dem das Produkt sicher und leistungsfähig ist
- Haltbarkeitsdauer (“Shelf Life”): Maximaler Zeitraum zwischen Produktion und Inbetriebnahme/Verwendung, in der das Produkt noch sicher und leistungsfähig ist
- Haltbarkeit/Stabilität (“Stability”): Produkteigenschaft, die angibt, dass das Produkt über einen Zeitraum des Produktlebenszyklus hinweg sicher und leistungsfähig bleibt
- Anbruchstabilität oder Haltbarkeit nach Anbruch (“In-Use Stability”): Zeitspanne, zwischen Inbetriebnahme/Verpackungsöffnung und Lebensende
- Ein Implantat könnte eine Lebensdauer von 15 Jahren haben (einschließlich der Zeit im menschlichen Körper), aber nur 2 Jahre Haltbarkeitsdauer (“shelf life”) bezogen auf die Sterilität innerhalb der Originalverpackung.
- Ein stoffliches Medizinprodukt könnte in der Originalverpackung drei Jahre gelagert werden (Haltbarkeitsdauer, “shelf life”), aber nach Anbruch innerhalb von 12 Monaten aufgebraucht werden müssen (Anbruchstabilität, “In-Use Stability”).
3.7.2 Tipps zum Umgang mit den Begriffen
- Verwenden Sie die Begriffe, die auch in den regulatorischen Anforderungen verwendet werden
- Greifen Sie die Begriffe in Ihrer Dokumentation auf. Legen Sie dort die Definitionen nochmal explizit in einem Glossar fest
- Geben Sie im Glossar die Referenz auf den Ursprung der Definition an oder vermerken Sie, dass es sich um Ihre eigene Definition handelt
- Machen Sie sich bewusst, dass es für Ihr Produkt mehrere Aspekte im Zusammenhang mit Alterung und Lebensdauer geben kann, die alle relevant sind. Entsprechende Nachweise sind dann für jeden einzelnen Aspekt notwendig.
3.7.3 Ergänzung: Stabilität von IVD-Reagenzien
Ganz besonders wichtig ist die Stabilität von Reagenzprodukten, inklusive Kalibrier- und Kontrollmaterialien für In-vitro-Diagnostika. Diese sind in der Regel sehr anfällig für Umwelteinflüsse wie beispielsweise Licht, Temperatur oder Erschütterung.
Die Stabilitätsprüfung muss nach Anhang II Kap. 6.3 der IVDR Teil der Technischen Dokumentation sein.
- Einschlägige Norm: ISO 23640 “Haltbarkeitsprüfung von Reagenzien für in-vitro-diagnostische Untersuchungen”
- Einschlägiges Guidance-Dokument: CLSI-Guidance-Dokument EP25-A
Die ISO 23640 wird voraussichtlich mit der IVDR harmonisiert. Die Norm wird ergänzt durch das CLSI-Guidance-Dokument EP25-A. Beide fordern die Echtzeitprüfung unter realistischen Bedingungen. Sie erlauben aber auch eine sogenannte beschleunigte Stabilitätsprüfung, um eine initiale Abschätzung der Stabilität bzw. Haltbarkeit eines Produkts zu ermöglichen.
4. Bestimmen der Lebensdauer
4.1 Bestimmen von Lebensdauer bei physischen Produkten
4.1.1 Allgemeine Kriterien für die Bestimmung der Lebensdauer
Leider geben rechtliche Regelungen wie MDR und IVDR auch keine direkten Hinweise darauf, wie die Lebensdauer eines Medizinproduktes bestimmt wird.
Um die Lebensdauer eines physischen Produktes zu bestimmen, sollten Hersteller sich daher an den einschlägigen Normen orientieren. Eine Norm, die alle Fälle abdeckt, gibt es jedoch nicht.
Die inzwischen überholte TR/ ISO 14969:2004, die sich noch auf die Anwendung der ISO 13485:2003 bezog, hatte eine sehr hilfreiche Zusammenfassung zur Festlegung der Lebensdauer von Medizinprodukten im Kapitel 7.1.3:
Entscheidungen hinsichtlich der Lebensdauer des Produkts können zum Teil erfolgen, um festgestellte Restrisiken zu steuern, die auf einen nicht vertretbaren Grad ansteigen können, wenn der Verwendungszeitraum eines Medizinprodukts ausgeweitet wird.
Auch Erwägungsgründe für die Festlegung der Lebensdauer beschrieb die Norm:
a) Lagerdauer des Medizinprodukts
b) Verfallsdatum von Medizinprodukten oder Bestandteilen, die im Laufe der Zeit Abbauvorgängen unterliegen
c) Anzahl der Zyklen oder Zeiträume der Verwendung des Medizinprodukts auf der Grundlage der Lebensdauerprüfung des Produkts
d) vorauszusehende Abbauvorgänge von Werkstoffen
e) Dauerhaftigkeit des Verpackungsmaterials
f) bei implantierbaren Produkten zum Restrisiko, das sich aus der gesamten Verweildauer des Produkts im Körper des Patienten ergibt
g) bei sterilen Medizinprodukten zur Fähigkeit, die Sterilität aufrechtzuerhalten
h) Fähigkeit bzw. Bereitwilligkeit der Organisation zu unterstützenden Dienstleistungen oder vertragliche oder durch Vorschriften gegebene Verpflichtungen zu diesen
i) Kosten und Verfügbarkeit von Ersatzteilen
j) juristische Erwägungen einschließlich der Haftbarkeit
k) Anzahl Nutzungen oder Betriebsstunden
Darüber hinaus gibt es auch kommerzielle Software-Angebote, mit denen sich die Lebensdauer modellieren lässt.
4.1.2 Spezielle Normen für einzelne Themengebiete
Aktuelle Normen gibt es für einzelne Themengebiete. Um diese zu identifizieren, empfiehlt sich eine ausführliche Recherche in Normendatenbanken, wie zum Beispiel die iso.org oder deren Volltextsuche. Beispiele für solche Normen sind:
- ISO 11346:2014 Elastomere oder thermoplastische Elastomere – Bestimmung der Lebensdauer und der höchsten Gebrauchstemperatur
- ISO 17526:2003 Optik und optische Instrumente – Laser und Laseranlagen – Lebensdauer von Lasern
- ISO/IEC 16963:2017 Information technology — Digitally recorded media for information interchange and storage — Test method for the estimation of lifetime of optical disks for long-term data storage
Für verschiedene Materialien gibt es auch Normen, die Alterungstests beschreiben. Nutzen Sie dazu die Volltextsuche der Normendatenbanken mit dem Begriff „accelerated aging test“.
Es gibt weitere Normen zu Alterungstests:
- ISO 188:2011 Elastomere oder thermoplastische Elastomere – Prüfung zur Bestimmung der beschleunigten Alterung und der Hitzebeständigkeit
- ISO 23640 Haltbarkeitsprüfung von Reagenzien für in-vitro-diagnostische Untersuchungen
Beachten Sie beim Festlegen der Lebensdauer unbedingt die in der erweiterten Zweckbestimmung des Produktes definierten Umgebungsbedingungen, speziell die Einflüsse durch Temperatur, Luftfeuchtigkeit, Luftdruck oder UV-Licht sowie die entsprechenden Lagerungs- und Transportbedingungen.
4.1.3 Wartung
Eventuell gibt es die Möglichkeit, gealterte Komponenten durch regelmäßige Prüfungen vor der Anwendung oder durch Selbsttests durch das Produkt zu untersuchen und so unsichere Zustände oder Leistungsverluste des Produktes rechtzeitig zu erkennen und entgegenzuwirken.
Mit Informationen zur Alterung und aus dem Risikomanagement können auch Wartungsstrategien entwickelt werden, die die festgesetzte Lebensdauer erhöhen können.
4.2 Bestimmen von Lebensdauer von Software-Produkten
Software selbst altert nicht. Doch die Umgebung der Software verändert sich: neue Betriebssysteme, neue Technologien, neue Kommunikationsprotokolle, etc.
Ein reines Softwareprodukt wird daher selten länger als einige Jahre Lebensdauer haben, ohne dass Modifikationen stattfinden. Daher wären fünf Jahre eine ganz gute Faustregel, wobei dies risikobasiert hergeleitet werden sollte.
Näheres erfahren Sie im Artikel „Lebensdauer von Software-Produkten“.
4.3 Sonstige Tipps zum Bestimmen von Lebensdauer
Im westlichen Raum gibt es zwar kaum Leitlinien dazu, worauf beim Bestimmen von Lebensdauer zu achten ist, doch ein Guidance-Dokument aus China hat einige Hinweise zusammengetragen. Daraus haben wir für Sie die folgenden Tipps zum Vorgehen beim Bestimmen von Lebensdauer abgeleitet:
1. Schritt: Produktlebensdauer in der Anforderungsspezifikation festlegen
Bereits vor bzw. zu Beginn der Entwicklung sollten Hersteller die Produktlebensdauer festlegen. Anhaltspunkte, wie lang dieser Zeitraum sein sollte, ergeben sich v. a. aus den folgenden Erwägungen:
- Kommerzielle Erwägungen: Vergleich mit dem Wettbewerb, Kundenwünsche, Rentabilität etc.
- Technische Erwägungen: Machbarkeit und Zeit bis zum Verschleiß von Bauteilen
- Empirische Daten: Erfahrungswerte aus der bisherigen Marktbeobachtung und aus Testergebnissen
Die Lebensdauer ist als Teil der Stakeholderanforderungen eine Vorgabe und kein Ergebnis der Entwicklung. Das Produktmanagement bringt vielmehr mit den Stakeholderanforderungen einen Vorschlag zur Lebensdauer ein. Über Risikomanagement, technische Datenblätter oder empirische Tests bewertet das Entwicklerteam diesen Vorschlag zur Lebensdauer und bestätigt ihn, zeigt Probleme auf oder bietet Optionen zur Lösung an (z. B. präventive Wartung, teurere Komponenten etc.).
2. Schritt: System- und Komponentenarchitektur erstellen
Mit den zuvor festgelegten Anforderungsspezifikationen im Hinterkopf erfolgt die Produktarchitektur. Hierbei müssen u. a. die passenden Technologien und Produktkomponenten identifiziert werden, die die nötigen Anforderungen erfüllen, um die Lebensdauer und alle anderen Anforderungen an das Produkt zu erfüllen.
In der Architektur sollten die Berechnungen und Herleitungen zur Auswahl/Auslegung bestimmter Komponenten nachvollziehbar enthalten sein. Ein Prüfer würde auf der Basis der Berechnungen entscheiden, ob bestimmte Tests durchgeführt werden müssen. Eine gute Dokumentation hilft somit auch den Prüfumfang einzugrenzen.
Die folgenden Aspekte sollten in der Architekturdokumentation enthalten sein:
- Herleitung der Lebensdauer
- Liste der kritischen Komponenten
- Zuverlässigkeitsberechnungen oder Ergebnisse von Simulationen
- Berechnungen von Sicherheitsfaktoren gemäß Tabelle 21 in der IEC 60601-1
- Umgebungsbedingungen mit Einfluss auf die Lebensdauer
- Ergebnisse aus der Risikoanalyse
- Ggf. Testanforderungen für den Nachweis der Lebensdauer
- Vergleich mit Referenzdesigns
- Normen, die betrachtet werden können
3. Schritt: Risiken im Zusammenhang mit Lebensdauer beherrschen
Im Risikomanagement müssen Hersteller auf Produkt- und Komponentenebene die Risiken identifizieren, die mit Sicherheit- und Leistungsfähigkeit über die Lebensdauer der Komponente hinweg verknüpft sind.
Dabei sind vor allem die folgenden Punkte zu bedenken:
- Produkt selbst berücksichtigen z. B. Reibung zwischen zwei beweglichen Bauteilen, Kraftwirkungen
- Anwendungskriterien berücksichtigen z. B. Häufigkeit und Intensität der Nutzung, Wartung und Reparatur
- Umgebung berücksichtigen z. B. Transport, Lagerung, Betriebsklima (Temperatur/Luftfeuchte, UV-Licht)
- Reinigung und Desinfektion beachten
- Die Kombinationen verschiedener Einflussfaktoren berücksichtigen
Reinigung und Desinfektion
Reinigung und Desinfektion sind Faktoren, die häufig bei der Lebensdauer übersehen werden. Die Verwendung des Produkts kann Reinigung und Desinfektion beinhalten. Die kumulative Wirkung von Erwärmungs- oder Trocknungsverfahren und chemischen Rückständen aus diesem Prozess kann die Leistung des Produkts beeinträchtigen und sollte hinsichtlich ihrer Auswirkungen auf die Lebensdauer des Produkts bewertet werden.
Verschiedene Sterilisationsmethoden (Dampfsterilisation, Ethylenoxidsterilisation, Strahlensterilisation usw.) und Verpackungsmethoden können sich unterschiedlich auf die Lebensdauer des Produkts auswirken. So sollten beispielsweise die Auswirkungen des Sterilisationsverfahrens (Methoden und Parameter) auf die Lebensdauer des Produkts berücksichtigt werden.
Optionen, um Lebensdauer zu verlängern
Für jede Komponente müssen Materialien und Funktionsprinzipien die geforderte Lebensdauer unterstützen. Sollte das nicht möglich sein, bestehen die folgenden Optionen, um die Lebensdauer der Komponente zu gewährleisten und Risiken zu minimieren:
- Option 1: Wartung
Hersteller können einen Präventivaustausch der Komponente vorsehen. Hierbei müssen dann die entsprechenden Wartungsintervalle in die Gebrauchsanweisung (IFU) einfließen. - Option 2: Prüffunktion
Alternativ (oder zusätzlich) können Hersteller eine Prüffunktion im Produkt implementieren, über die der Ausfall von Komponenten (oder sonstige Einschränkungen der Sicherheit- und Leistungsfähigkeit) zuverlässig erkannt wird. Dies ist immer dann eine Option, wenn durch die Prüffunktion das Produkt in einen sicheren Zustand versetzen wird. Das bedeutet aber auch, dass diese Option nicht bei lebenserhaltenden/-unterstützenden Systemen greift. Ist deren Leistung eingeschränkt oder fallen diese gar aus und die Prüffunktion wird getriggert, ist es bereits zu spät. - Option 3: Produktlebensdauer reduzieren
Sollte keine der anderen Optionen infrage kommen, können Hersteller nur noch die Produktlebensdauer auf eine kürzere Lebensdauer reduzieren und dies bei der Anforderungsspezifikation anpassen.
5. Regulatorische Anforderungen an Lebensdauer
Es gibt keine rechtliche Regelung, die sich ausschließlich und abschließend mit der Lebensdauer von Medizinprodukten befasst. Vielmehr werden stets einzelne Aspekte geregelt. Dazu zählen insbesondere:
- Neuaufbereitung und Auswirkung auf die Lebensdauer
- Kennzeichnung von Lebensdauer
- Qualitätsmanagement-System und Lebensdauer
- Kommunikation der Lebensdauer
- Aktive Medizinprodukte und Lebensdauer
5.1 Neuaufbereitung lässt Lebensdauer neu beginnen
„Neuaufbereitung“ im Sinne der Herstellerdefinition bezeichnet die vollständige Rekonstruktion eines bereits in Verkehr gebrachten oder in Betrieb genommenen Produkts oder die Herstellung eines neuen Produkts aus gebrauchten Produkten mit dem Ziel, dass das Produkt den Anforderungen dieser Verordnung entspricht; dabei beginnt für die als neu aufbereiteten Produkte eine neue Lebensdauer
Quelle: Art. 2 Nr. 31 MDR
Demnach können Medizinprodukte wieder in einen Zustand versetzt werden, der entweder dem ursprünglichen eines entsprechenden neuwertigen Produktes entspricht oder in einen Zustand, für den eine neue Lebensdauer festzulegen ist.
Wie diese Rekonstruktion aussieht, muss der Hersteller jeweils selbst festlegen und dokumentieren. Eventuell sind auch Aspekte der Reinigung, Desinfektion und Sterilisierung zu beachten. Hilfestellung dabei kann die ISO 17664 geben.
Eine Entsorgung des Medizinproduktes könnte also durch eine Neuaufbereitung verhindert werden. Ob diese wirtschaftlich Sinn ergibt, muss der Betreiber oder der Hersteller entscheiden.
Hierbei sind Betreiber in der Regel auf die Expertise des Herstellers angewiesen, um den Umfang, die Methoden und Details einer Neuaufbereitung zu wählen. Allerdings besteht zur Beratung keine rechtliche Verpflichtung, was für Betreiber eine Herausforderung darstellt.
5.2 Lebensdauer muss in Begleitdokumenten genannt sein
Für implantierbare Produkte legt die MDR fest:
(1) Der Hersteller eines implantierbaren Produkts liefert zusammen mit dem Produkt folgende Informationen: […]
Quelle: Art. 18 MDR
c) Angaben zur voraussichtlichen Lebensdauer des Produkts und zu den notwendigen Folgemaßnahmen
Dies ist auf Ebene der europäischen Regelungen die einzige Stelle, an der die Angabe der Lebensdauer in den Begleitinformationen eines Produktes explizit gefordert ist. Allerdings befassen sich noch weitere Regelungen der MDR mit Angaben in den Begleitdokumenten in Bezug auf Lebensdauer.
Anhang I MDR, Abschnitt 23.4: Angaben in der Gebrauchsanweisung
k) alle Angaben, mit denen überprüft werden kann, ob das Produkt ordnungsgemäß installiert wurde und für den sicheren und vom Hersteller beabsichtigten Betrieb bereit ist, sowie gegebenenfalls
[…]
— Angaben zu der möglicherweise erforderlichen Kalibrierung, mit der der ordnungsgemäße und sichere Betrieb des Produkts während seiner erwarteten Lebensdauer gewährleistet wird, und […]
Neben dem Austausch von Verschleißteilen und der regelmäßigen Überprüfung von Medizinprodukten im Rahmen der Wartung, spielen also auch Kalibrierungen eine Rolle, die der Hersteller definieren muss.
Anhang VI MDR, Teil B Das UDI-System
4.10. Wiederverwendbare Produkte tragen den UDI-Träger auf dem Produkt selbst. Der UDI-Träger für wiederverwendbare Produkte, bei denen zwischen den Anwendungen am Patienten eine Reinigung, Desinfektion, Sterilisation oder Aufbereitung erforderlich ist, muss dauerhaft angebracht und nach jedem Verfahren, das zur Vorbereitung des Produkts für die nachfolgende Verwendung durchgeführt wird, während der gesamten erwarteten Lebensdauer des Produkts lesbar sein.
4.11. Der UDI-Träger muss bei normaler Anwendung während der erwarteten Lebensdauer des Produkts lesbar sein.
Dieser Punkt handelt zwar von der Kennzeichnung des Produktes, bezieht sich aber eher auf die Auslegung der Kennzeichnung. Es wird gefordert, dass der Hersteller nachweisen kann, dass das Typenschild mit UDI dauerhaft am Produkt lesbar bleibt und allen äußeren Einflüssen gemäß bestimmungsgemäßem Gebrauch über die gesamte Lebenszeit standhält. Die Auswahl des Materials der Kennzeichnung spielt dabei genauso eine Rolle, wie Druckfarbe und -technik, Licht- und Temperatureinflüsse, Flüssigkeiten und Luftfeuchte, Vibrationen und mechanische Belastungen oder die Angabe geeigneter Desinfektionsmittel.
Bei der dauerhaften Lesbarkeit von Typenschildern ist auch darauf zu achten, dass der Austausch von Komponenten des Produktes, auf denen das Typenschild angebracht ist, technisch und prozessual vorgesehen ist.
Sind Techniker im Feld entsprechend sensibilisiert? Können neue Typenschilder mit den ursprünglichen Daten eines Produktes bei Bedarf generiert werden? Können sie im Feld angebracht werden? Wie werden sie dabei verifiziert? Gerade diese scheinbar belanglosen Sonderfälle reißen leicht Lücken in die sonst einwandfreie regulatorische Konformität.
5.3 Lebensdauer bestimmt die PMS und PMCF
Artikel 83 MDR: Überwachung nach dem Inverkehrbringen
Die MDR stellt Anforderungen an die Post-Market Surveillance (PMS), die auf deutsch als “Überwachung nach dem Inverkehrbringen“ bezeichnet wird:
(2) Das System zur Überwachung nach dem Inverkehrbringen ist geeignet, aktiv und systematisch einschlägige Daten über die Qualität, die Leistung und die Sicherheit eines Produkts während dessen gesamter Lebensdauer zu sammeln, aufzuzeichnen und zu analysieren sowie die erforderlichen Schlussfolgerungen zu ziehen und etwaige Präventiv- oder Korrekturmaßnahmen zu ermitteln, durchzuführen und zu überwachen.
Hier scheint sich die Lebensdauer nicht nur auf das Einzelprodukt zu beziehen, sondern auf alle im Markt befindlichen Produkte. Wichtig ist dieser Punkt insbesondere dann, wenn der Hersteller das Produkt bereits abgekündigt hat (siehe „end of life“), es aber weiterhin im Markt überwachen muss. Nämlich genau so lange, bis das letzte Produkt im Markt seine angegebene Lebensdauer erreicht hat. Wenn nachweislich kein Produkt mehr im Markt ist, kann auch die Überwachung nach dem Inverkehrbringen enden.
Artikel 86 MDR: Regelmäßig aktualisierter Bericht über die Sicherheit
Während der gesamten Lebensdauer des betreffenden Produkts wird in diesem Sicherheitsbericht Folgendes aufgeführt: […]
Auch dies gilt für alle im Markt befindlichen Produkte und beschreibt die Pflicht, bestimmte Informationen zu sammeln, auszuwerten und in einem regelmäßigen Bericht zusammenzufassen.
Anhang XIV MDR: Klinische Nachbeobachtung
Zudem fordert die MDR eine klinische Nachbeobachtung, die Post-Markt Clinical Follow-up (PMCF). Auch diese hängt von der Lebensdauer ab:
5. Bei der klinischen Nachbeobachtung nach dem Inverkehrbringen sammelt und bewertet der Hersteller auf proaktive Weise klinische Daten, die aus der Verwendung eines die CE-Kennzeichnung tragenden, im Rahmen seiner Zweckbestimmung gemäß dem einschlägigen Konformitätsbewertungsverfahren in den Verkehr gebrachten oder in Betrieb genommenen Produkts im oder am menschlichen Körper hervorgehen, um die Sicherheit und die Leistung während der erwarteten Lebensdauer des Produkts zu bestätigen, die fortwährende Vertretbarkeit der ermittelten Risiken zu gewährleisten und auf der Grundlage sachdienlicher Belege neu entstehende Risiken zu erkennen.
Da Lebensdauer definitionsgemäß die Zeitspanne ist, in der ein Medizinprodukt sicher und leistungsfähig betrieben werden kann, ist es notwendig, hierüber ausreichend Daten zu sammeln. Damit lässt sich für den Hersteller der Beweis führen, dass alle seine Annahmen und Angaben hierzu berechtigt waren.
5.4 Kommunikation der Lebensdauer
Anwender und Betreiber müssen wissen, ab wann sie das Produkt nicht mehr betreiben dürfen, weil Sicherheit und Leistungsfähigkeit nicht mehr garantiert sind. Schon daraus ergibt sich die logische Notwendigkeit zur Kommunikation der Lebensdauer durch den Hersteller.
Doch wie sieht es mit verbindlichen gesetzlichen Anforderungen dazu aus?
MDR
Tatsächlich fordert die MDR in Artikel 18 (1)(c) eine verbindliche Angabe der Lebensdauer nur im Zusammenhang mit implantierbaren Produkten (siehe oben).
IMDRF
Das IMDRF-Dokument „GHTF SG2 N21 R8 May 1999“ führt in Kapitel 2.3 auf:
The service life must be specified by the device manufacturer and included in the master record [technical file] or, where appropriate, the instructions for use (IFU).
“Where appropriate” kann man dabei nur so verstehen, dass es auch Produkte mit praktisch unbegrenzter Lebensdauer geben kann, für die eine solche Angabe keinen Sinn ergibt. Genauso für sterile Einmalprodukte, bei denen eher die Haltbarkeitsdauer relevant ist, nicht die Lebensdauer, da sie meist unmittelbar angewendet und anschließend entsorgt werden.
IEC 60601-1-11:2015
Die IEC 60601-1-11:2015 fordert explizit die Angabe der erwarteten Betriebs-Lebensdauer in den Begleitpapieren von medizinisch elektrischen Geräten zur Verwendung in häuslicher Umgebung.
Insgesamt lässt sich also festhalten, dass die Angabe der Lebensdauer eines Medizinproduktes in den Begleitpapieren in den meisten Fällen dringend notwendig ist, teilweise gesetzlich oder regulatorisch sogar gefordert wird. In jedem Fall muss sie Teil der Technischen Dokumentation sein.
5.5 Anforderungen an aktive Medizinprodukte nach IEC 60601
In der IEC 60601-1 ergibt sich die Relevanz von Lebensdauer in Zusammenhang mit dem Begriff “Erstfehlersicherheit”. In der IEC 60601-1 ist die Erstfehlersicherheit ein wichtiges Konzept.
Merkmal eines ME-Geräts oder von Teilen des ME-Geräts, wodurch es während der zu erwartenden Betriebs-Lebensdauer beim ersten Fehler frei von unvertretbaren Risiken bleibt
Diese Forderung können Hersteller nur dann erfüllen, wenn sie die Lebensdauer festgelegt haben und sie über empirische Tests und das Risikomanagement entsprechend nachvollziehbar belegen können.
Und tatsächlich richtet die IEC 60601-1 in ihrer Edition 3.1 im Anhang A den Appell an die Hersteller:
In den Begleitpapieren sollte der Hersteller Informationen geben, die es der verantwortlichen Organisation erlauben abzuschätzen, wann das ME-Gerät sich dem Ende seiner Lebensdauer nähert. Diese Informationen sollten die zu erwartende Betriebs-Lebensdauer enthalten, wie sie vom Hersteller festgelegt wurde (z. B. in Betriebsjahren oder in Anzahl von Benutzungen), aber sie könnten auch Prüfungen enthalten, die als Teil der vorsorglichen Wartung durchgeführt werden, oder andere Kriterien, die es der verantwortlichen Organisation erlauben, eine angemessene Entscheidung zu treffen. Der Bedarf für derartige Informationen und die angemessene Weise, wie sie präsentiert werden, sollte als Teil des Risikomanagement-Prozess behandelt werden.
5.6 Anforderungen in China
Besondere Überraschungen erleben Hersteller derzeit bei der Zulassung ihrer Produkte in China. Dort spielt die Lebensdauer nämlich eine so große Rolle, dass es sogar eine eigene NMPA-Guidance dafür gibt. Sinngemäß übersetzt mit
“Richtlinie für die technische Überprüfung der Lebensdauer für aktive Medizinprodukte (Nr. 23 von 2019)”
Im Rahmen der Registrierung von Medizinprodukten müssen Antragsteller die in den Begleitpapieren angegebene Lebensdauer ihrer Produkte mit Nachweisen belegen.
Das Regelwerk beschreibt im Detail, wie die Lebensdauer bestimmt wird und wie sie im Risikomanagement Berücksichtigung findet.
Im Grunde deckt sich dies mit den regulatorischen Anforderungen in Europa, wird hier aber im Gegensatz zu China wesentlich seltener Angriffspunkt im Zulassungsverfahren.
6. Fazit
Sicherheit und Leistungsfähigkeit eines Medizinproduktes sind nur während der vorgegebenen Lebensdauer gegeben. Bei deren Bestimmung sind alle risikorelevanten Punkte zu berücksichtigen. Unglücklicherweise nehmen jedoch viele Hersteller und Betreiber das Konzept nicht ganz so genau, u. a., weil eine längere Lebensdauer ökonomisch von Vorteil sein kann und die Definitionen unscharf sind.
Begriffe wie Lebensdauer, Haltbarkeitsdauer, Service-Zeit, Betriebszeit und “end of life” sind keine Synonyme. Sie müssen aber genau verstanden sein, um die regulatorischen Anforderungen zu erfüllen.
In Europa spielt die Lebensdauer in den einschlägigen Dokumenten, wie der Risikomanagement-Akte und der Testdokumentation, eine große Rolle. Die IEC 60601-1 gibt hierbei wertvolle Hinweise zur konkreten Umsetzung.
Auch Märkte wie China fordern bei der Produktzulassung nicht nur die Angabe der Lebensdauer in den Begleitpapieren, sondern auch ihre Herleitung und belastbare Nachweise dazu in der Technischen Dokumentation.
Hersteller in Europa tun daher gut daran, in Zukunft die gewünschte Lebensdauer als Design Input festzulegen. Sie sollten die tatsächliche Lebensdauer ihrer Produkte als “wesentliches Entwicklungsergebnis” (Essential Design Output im Sinne des 21 CFR 820.30d) viel stringenter im Lebenszyklusprozess berücksichtigen und ausreichend Nachweise für deren Angabe liefern.
Die Expertinnen und Experten des Johner Instituts unterstützen Sie gern bei Fragen zur Lebensdauer von Medizinprodukten. Kontaktieren Sie uns einfach über das Formular oder schreiben Sie eine E-Mail.
Änderungshistorie:
- 2025-03-23: Grafik in Abschnitt 1 eingefügt, Nummerierung geändert, Definitionen ergänzt. Kapitel 3.6 eingefügt.
- 2022-08-29: Unter 4c) Tipps zum Bestimmen von Lebensdauer ergänzt


Hervorragender Übersichtsartikel!
Vielen Dank für diese großartige Rückmeldung, Herr Müller! Das bestärkt uns in unserer Arbeit und motiviert uns für weitere Beiträge.
Herzliche Grüße
Christian Rosenzweig
Danke für die guten und interessanten Beiträge.
h/p/cosmos hat zum Thema End of Life und den damit verbundenen Haftungsrisken für Betreiber und Hersteller beim Fachanwalt Dr. Lücker eine Expertenmeinung beauftragt.
https://www.hpcosmos.com/de/aktuelles/medizinprodukte-lebensdauer-end-life-altgeraete
Herzlichen Dank, Herr Harrer, für diese wichtige Ergänzung. Tatsächlich scheinen Publikationen zu einem juristischen Konsens im Moment noch rar zu sein. Umso mehr freue ich mich, dass Sie hier eine juristische Stellungnahme verlinken.
Freundlichste Grüße
Christian Rosenzweig
Sehr gute Sammlung von Theorie.
Was soll der Hersteller die Lebensdauer definieren?
Evaluation der kritischen Komponente, Real time test oder Aging test.
Wie anders ist die Lebensdauer bei Initial – und Change-Registrierung?
Und wie meistert man die Analyse und Test, wie oft?
Vielen Dank für Ihren Kommentar, Christian. Wir werden das Thema der Bestimmung der Lebensdauer in einem weiteren Blogbeitrag nochmal aufgreifen.
Herzliche Grüße
Christian Rosenzweig
Vielen Dank für diesen hervorragenden Beitrag.
Besonders Interessant bzw.in anderen Puplikationen weitgehend vernachlässigt waren die regulatorischen Unterschiede im asiatischen Raum .
Weiter so!
Besten Dank für Ihre Wertschätzung, Herr Weiss!
Ergänzend zum Artikel hier ein über die Behörde gemeldeter Rückruf:
https://www.bfarm.de/SharedDocs/Kundeninfos/DE/14/2022/08054-22_kundeninfo_de.html
In der Gebrauchsanweisung hatte die Angabe der Lebensdauer gefehlt. Einmal mehr ein Hinweis darauf, wie wichtig diese Angabe für die Anwender tatsächlich ist!
Sehr geehrter Herr Prof. Dr. Johner,
dürfen Medizinprodukte, die abgelaufen sind (shelf-life) an Kunden verschenkt werden, wenn jenen die Sachlage bekannt ist?
Freundliche Grüße!
C. Nestmann
Liebe Frau Nestmann,
wenn Sie offiziell Produkte mit abgelaufener Shelf-Life in Verkehr bringen, wären die Produkte „nicht-konform“ im Markt bereitgestellt, denn nach Ablauf der Shelf-Life wäre deren Sterilität möglicherweise nicht mehr gegeben. Eine mündliche oder schriftliche Stellungnahme dazu nutzt Ihnen auch nichts, denn Sie wissen nicht, wer die Produkte in die Hände bekommt und was damit passiert. Um sich juristisch auf sicherem Boden zu bewegen, würde ich das Aufbringen eines prominenten Labels „Not for clinical use“ auf jedem einzelnen betroffenen Produkt empfehlen (in Warnfarben). Dann kann das Produkt zwar noch zu Trainings- und Anschauungszwecken verteilt werden, aber nicht mehr im klinischen Kontext angewendet werden.
Herzliche Grüße
Christian Rosenzweig
Sehr geehrter Herr Rosenzweig
Vielen Dank für den tollen Beitrag!
Gibt es eine Norm oder Richtlinie, die sich mit der wiederverwendbarkeit von Instrumenten und der damit verundenen Funktionskontrolle beschäftigt? Anleitungen für Das Fachpersonal der Sterilisation sind meist ziemlich offen formuliert. Als Beispiel wird in einem Dokument für die Funktionskontrolle erwähnt, dass ein Instrument, falls es zu tiefe Kratzer aufweist, nicht mehr verwendet werden darf. Wie tief diese Kratzer sein dürfen, bleibt aber offen.
Freundliche Grüße!
M. Baumann
Liebe Frau Baumann,
Vielen Dank für Ihre wertvolle Rückmeldung und die weitere Vertiefung des Themas!
Es gibt eine Reihe allgemeiner Vorgaben. Hier eine Auswahl:
– ISO 17664
– Themenwebseite der EU
– Informationen vom Robert-Koch-Institut in Deutschland
Diese Vorgaben sind natürlich, wie Sie schon sagen, allgemein gehalten und nicht spezifisch für das individuelle Medizinprodukt. Im Einzelfall kommt es also darauf an, welche spezifischen Anforderungen an das Medizinprodukt gestellt werden und was dessen sicherheitsbezogene Merkmale sind. Das weiß in der Regel nur der Hersteller des Produktes genau, weil er es in seinem Risikomanagementprozess darstellen muss. Wenn er die Wiederaufbereitung unterstützt, stellt er die entsprechenden Informationen zur Verfügung.
Herzliche Grüße
Christian Rosenzweig
Bei wiederverwendbaren (chir.) Instrumenten (Instrumentensets) war es in der Vergangenheit üblich (und akzeptiert) keine Lebensdauer anzugeben. Es wurde aber in den Aufbereitungsanweisungen sinngemäss folgendes geschrieben:
Die Lebensdauer wird durch die Anwendungen und Aufbereitungen nur unwesentlich beeinflusst. Sie hängt eher ab von den Anwendungen und dem Umgang mit den Instrumenten. Deshalb muss bei jeder Aufbereitung die Funktion die Funktion geprüft werden, und ggf. ist bei Abnutzung oder Defekten die Lebensdauer dann erreicht.
Wenn jetzt (MDR) für jedes Instrument eine Lebensdauer festgelegt werden muss (z.B. 2 Jahre oder 100 Aufbereitungen), würde das dazu führen, dass noch voll intakte Instrumente nach Ablauf der Lebensdauer, nicht mehr verwendet werden dürften und ersetzt werden müssten (immense Kosten!). Obwohl die Sicherheit auch weiterhin über die Prüfungen bei jeder Aufbereitung sichergestellt wäre.
(Auch erfolgt eine Erfassung der Anzahl von Aufbereitungen jedes einzelnen Instruments, nach meinem Kenntnisstand, aktuell praktisch in keiner Klinik).
Gibt es Erfahrungen, wie die Benannten Stellen diese Problematik behandeln/ob weiterhin dieses Vorgehen (keine Angabe einer fixen Lebensdauer) möglich wäre?
Viele Grüsse
Lieber Herr Schneider,
ein wichtiger Gedanke, den Sie da einbringen. Tatsächlich kenne ich solche Diskussionen auch aus der Praxis.
Die Definition von Lebensdauer lautet „Die Zeitspanne, in der das Medizinprodukt sicher und leistungsfähig betrieben werden kann, unter Einhaltung der vom Hersteller vorgegebenen Wartungen und Prüfungen“. Wenn Sie sagen, dass man dem Produkt ansehen kann, ob es noch funktionstüchtig ist, dann könnte der Hersteller auch Vorgaben für eine solche Prüfung machen, die zum Aussondern oder Weiterverwenden des Produktes bis zum endgültigen Lebensende führen. Das endgültige Lebensende würde der Hersteller aufgrund seiner eigenen Lebensdauer-Tests festlegen bzw. belegen.
Im Übrigen führt das Ende der angegebenen Lebensdauer des Medizinproduktes zum Übergang der Verantwortung für Sicherheit und Leistungsfähigkeit vom Hersteller auf den Betreiber. Falls dieser also selbst die Sicherheit und Leistungsfähigkeit über das Lebenszeitende hinaus belegen kann, könnte er das Produkt auch weiter verwenden. Dazu rate ich aber nicht, denn nur der ursprüngliche Hersteller hat die vollen Informationen zur Auslegung und den Risiken des Produktes.
Im Moment werden immer wieder Nachhaltigkeitsdebatten geführt, auch im Bereich der Medizinprodukte. Gerade die EU mit ihren entsprechenden politischen Programmen geht hier gute Wege. Es ist also absehbar, dass neben er Sicherheit und Leistungsfähigkeit der Produkte auch deren Wiederverwendung oder Auslegung für eine lange Lebensdauer mit berücksichtigt werden müssen. Natürlich auch wieder unter strenger Berücksichtigung von Sicherheit und Leistungsfähigkeit des Produktes.
Herzliche Grüße
Christian Rosenzweig
Sehr geehrter Herr Rosenzweig,
sehr interessante Artikel, vielen Dank dafür.
Gibt es Vorgaben in Bezug auf die Haltbarkeitsdauer eines sterilen Medizinproduktes wie viele Monate/Jahre (?) diese bei Auslieferung vom Hersteller an den Händler mindestens betragen muss und wie viele Monate/Jahre wenn der Händler dieses Produkt wiederum an den Endkunden ausliefert?
Herzliche Grüße C. Kunkel
Liebe Frau Kunkel, eine sehr interessante Frage! Mir sind solche regulatorischen Forderungen nicht bekannt. Meines Wissens legt allein der Medizinproduktehersteller die Haltbarkeitsdauer fest. Ich gehe davon aus, dass eher der Markt regelt, ob Medizinprodukte mit zu kurzer Haltbarkeitsdauer wettbewerbsfähig sind. Die Länge der Haltbarkeitsdauer wiederum ergibt sich aus der Güte der Sterilbarrieren, die nach Stand der Technik eben nur eine begrenzte Haltbarkeitsdauer ermöglichen.
Herzliche Grüße
Christian Rosenzweig
Lieber Herr Rosenzweig,
vielen Dank für die schnelle Rückmeldung.
Wenn ich Sie korrekt verstehe, gibt es hierzu für den deutschen Markt keine gesetzlichen Vorgaben, so das jeder Händler hier seinen individuellen Rahmen innerhalb der Haltbarkeit seiner Produkte festlegen kann, solange die Ware innerhalb dieser Frist beim Endkunden ankommt?
Letztlich müsste dabei aber auch sicher gestellt sein, dass der Kunde eine realistische Chance hat, diese Ware innerhalb der Frist zu verbrauchen.
Ich stelle mir bspw. eine Quartalslieferung eines Hilfsmittels vor – dann müsste entsprechend die Haltbarkeit berechnet sein auf Lieferdatum + 3 Monate?
Herzliche Grüße C. Kunkel
Sie haben völlig recht, Frau Kunkel. Die Ware muss natürlich so beim Kunden ankommen, dass er sie ausreichend bevorraten und innerhalb der Haltbarkeitszeitspanne verbrauchen kann.
Es gibt auch noch eine schöne Zusammenfassung der Händlerpflichten unter dieser Quelle: https://www.dgihv.org/wp-content/uploads/DGIHV_MDR_Leitfaden_H%C3%A4ndler.pdf
Lieber Herr Rosenzweig,
ich hätte eine Frage: Ist für die Bestimmung der Lagerstabilität bei nicht sterilen Medizinprodukten zwingend eine Echtzeitlagerung notwendig, wenn die propagierte Lagerstabilität 2 Jahre nicht überschreitet?
Muss man immer eine Echtzeitlagerung anstoßen?
VG
Liebe Frau Balolong,
meines Wissens ist es bei den meisten Produkten typischerweise nicht explizit gefordert, einen Echtzeittest durchzuführen. Beschleunigte Alterung, z.B. über eine Klimakammer, wären demnach bei ausreichender Herleitung und Begründung der Testspezifikation auch möglich. Ich könnte mir aber vorstellen, dass es in manchen Märkten für manche spezielle Produkte individuelle Normen oder Leitfäden gibt, die anzuwenden sind.
Herzliche Grüße
Christian Rosenzweig
Lieber Herr Rosenzweig,
Welche Maßnahmen muss der Hersteller bei einer Verlängerung der Haltbarkeit ergreifen, wenn das Produkt bereits bei Händlern/Endkunden ist?
Reicht z.B. eine Erklärung über die verlängerte Haltbarkeit und ein aktualisiertes Produktdatenblatt?
Gibt es dazu bestimmte gesetzliche Anforderungen in der IVDR?
Herzliche Grüße
Ich habe Ihre Frage so verstanden, dass Sie im Nachhinein die Angabe zur Haltbarkeit in der Kennzeichnung Ihres Produktes korrigieren möchten. Nämlich aufgrund neuer Erkenntnisse auf eine längere Haltbarkeit.
Im Sinne der Regulatorik ist das zunächst ein unkritischer Fall, weil Sicherheit und Leistungsfähigkeit der Produkte nicht betroffen wären. Es geht wohl eher um die Nachhaltigkeit, weil Produkte aufgrund der falsch niedrigen Haltbarkeitsdauer eventuell unnötig unbenutzt entsorgt werden würden.
Demnach liegt kein meldepflichtiges Ereignis vor und damit müssen Sie die Anpassung der Kennzeichnung im Markt auch nicht als FSCA (Field Safety Corrective Action) durchführen. Es handelt sich also gemäß MDR/IVDR um eine Korrekturmaßnahme im Feld:
MDR, Artikel 2
67. „Korrekturmaßnahme“ bezeichnet eine Maßnahme zur Beseitigung der Ursache eines potenziellen oder vorhandenen Mangels an Konformität oder einer sonstigen unerwünschten Situation;
Sie ändern also Ihr Produktdesign über Ihre eigenen Vorgaben des Qualitätsmanagementsystems (unter einem Änderungs-Management-Prozess nach ISO 13485), geben diese Änderung intern frei und sorgen dann dafür, dass ihre Kunden informiert werden und angepasste Produkte/Kennzeichnungen erhalten (z.B. durch Rücksendung und Austausch). Eine reine Information per Brief oder Nachricht halte ich für etwas dünn, weil auf dem Produkt meist ein Haltbarkeitsdatum angegeben ist und der Kunde sich dann jeweils pro Produkt das neue Haltbarkeitsdatum individuell errechnen müsste. Die Fehler, die dabei passieren könnten, lägen wieder in Ihrer Verantwortung als Hersteller. Sorgen Sie also besser dafür, dass die Kennzeichnung auf dem und mit dem Produkt ausgetauscht werden. Denken Sie auch daran, dass eine Änderung an der Produkthaltbarkeit unter Umständen eine Mitteilung an Ihre Benannte Stelle notwendig machen könnte.
Bei Bedarf unterstützen unsere Berater:innen sehr gerne bei der ordnungsgemäßen Umsetzung.
Vielen Dank für den Beitrag. Ich frage mich gerade ob für Sterilprodukte die Lagerstabilität über eine beschleunigte Alterung nachgewiesen werden kann, oder ist zwigend eine Echtzeitalterungsvalidierung notwendig?
Vielen lieben Dank!
Liebe Mona,
die Lagerstabilität von Produkten, die sterilisiert werden, kann zunächst mit beschleunigter Alterung nachgewiesen werden, z.B. um das Produkt mit einer gewissen Haltbarkeitsdauer vor Abschluss der vollständigen Echtzeit-Validierung zu vertreiben, allerdings müssen diese Daten mit Studien zur Echtzeitalterung verifiziert werden. Das fordert die ISO 11607-1.
Die FDA benennt, „Products labeled as sterile are expected to be free from viable microbial contamination throughout the product’s entire shelf life“ (https://www.fda.gov/media/76338/download)
Der referenzierte Standard ASTM F1980-07 Standard Guide for Accelerated Aging of Sterile Barrier Systems for Medical Devices gilt auch für Medizinprodukte: https://www.astm.org/f1980-07.html
„This guide provides information for developing accelerated aging protocols to rapidly determine the effects, if any, due to the passage of time on the sterile integrity of the sterile barrier system (SBS), as defined in ANSI/AAMI/ISO 11607-1:2006 and the physical properties of their component packaging materials.“
FAZIT: Beschleunigte Alterungsstudien sind zunächst für eine initiale Produktfreigabe möglich, müssen aber produkt-spezifisch über das Risikomanagement begründet werden. Zudem müssen sinnvolle Akzeptanzkriterien gewählt werden. Spätere Echtzeit-Validierung ist erforderlich.
Herzliche Grüße
Christian Rosenzweig
Sehr geehrte Damen und Herren,
ist hier nicht ein Widerspruch gegeben?
(Ich verkürze den Inhalt etwas)
3.6 Nutzungsdauer
Nutzungsdauer = Dauer, in der das Produkt nach dessen Inbetriebnahme bzw. Öffnung bis zu dessen Lebensende genutzt werden kann. Nutzungsdauer = Synonym für Anwendungsdauer bzw. „Service-Life“.
3,7 Zwischenfazit und -zusammenfassung
3.7.1 Begriffe
Lebensdauer („Service Life“): Gesamte Zeitspanne von Produktion bis Lebensende des Produktes,…
Müsste bei 3.7.1. Lebensdauer dann nicht als „In-Use-Stability“ bezeichnet werden?
Vielen Dank generell für Ihre tollen Artikel!
Mit freundlichen Grüßen
Lieber Herr Mühe, vielen Dank für Ihren wertvollen Kommentar. Mir war beim Erstellen des Artikels schon bewusst, dass die Begriffe nicht in jedem Szenario in gleicher Weise greifen. Deshalb müsste man für jeden Anwendungsfall eine eigene Liste an Begriffen und Definitionen machen: für sterile Einmalprodukte, für stoffliche Produkte, für elektromedizinische Produkte, für Implantate, etc.
Der Begriff Anbruchstabilität (“in-use stability”) wird eben nur bei manchen Produkten (z.B. dentale Füllmaterialien) so bezeichnet.
Herzliche Grüße
Christian Rosenzweig
Lieber Herr Rosenzweig,
wäre es, als Instandhalter, aus regulatorischer und sicherheitstechnischer Sicht zulässig, bei einem Anwendungsteil eines Medizinprodukts, das nur für eine begrenzte Anzahl an Anwendungen vorgesehen ist, den Anwendungscounter zurückzusetzen – nachdem eine umfassende Analyse des Geräts durchgeführt wurde?
Die Analyse würde durch eine gründliche Sichtkontrolle auf mechanische Beschädigungen, Abnutzung oder Materialveränderungen, eine vollständige Funktionsprüfung gemäß Produktspezifikation sowie eine detaillierte elektronische Überprüfung aller relevanten technischen Parameter sicherstellen, dass Sicherheit und Leistungsfähigkeit des Geräts uneingeschränkt gegeben sind.
Lieber Herr Tschaikner, das ist eine interessante Frage. Sie meinen mit „Instandhalter“ einen Betreiber oder Dienstleister des Betreibers? Der Hersteller des Produktes hat über sein Risikomanagement und seine Nachweisführung zur Lebensdauer die Verantwortung für die Produktlebensdauer übernommen (genau so fordert es die Regulatorik). Als Instandhalter kennen Sie das Produkt meistens nicht gut genug, um beurteilen zu können, ob es nach Ablauf der Lebensdauer noch sicher und leistungsfähig genug ist. Sie können z.B. durch Sichtkontrollen nicht die biologische Sicherheit bewerten, oder Materialermüdung erkennen. Formal kann aber der Betreiber sagen, dass er das Produkt trotz gegensätzlicher Vorgaben des Herstellers über die Lebensdauer hinweg verwendet. Wenn dann etwas passiert (Produktsicherheit), haftet jedoch vollständig der Betreiber, nicht der Hersteller.
Um eine ähnliche Problematik geht es übrigens auch im Artikel 23 der MDR („Teile und Komponenten“), wo Drittanbieter voll in die Haftung genommen werden.
Ich hoffe, das hilft Ihnen für Ihre Fragestellung weiter.
Herzliche Grüße
Christian Rosenzweig
Lieber Herr Rosenzweig,
vielen Dank für Ihre Einschätzung.
Wenn ein Instandhalter (oder Reparateur) den Anwendungscounter zurücksetzt, wird damit faktisch die Lebensdauer des Produkts über den vom Hersteller festgelegten Zeitraum hinaus verlängert – und die Verantwortung für die sichere Weiternutzung übernommen.
Die Frage ist nun: Begründet eine solche Maßnahme bereits eine Herstellerrolle im Sinne der MDR, mit allen damit verbundenen Pflichten? Oder bewegt man sich hierbei noch im Rahmen von Artikel 23 MDR?
Reicht es also aus, wenn auf Basis einer geeigneten und dokumentierten technischen Prüfung (inklusive Risikomanagement) festgestellt wird, dass das Risiko vertretbar ist und das Produkt weiterhin sicher betrieben werden kann?
Vielen Dank im Voraus.
Mit freundlichen Grüßen
Liebe Ursula, jetzt verlassen wir langsam den Bereich der regulatorischen Interpretation und bewegen uns stark im rechtlichen Interpretations-Spielraum. Ich kann Ihnen leider keine Rechtsberatung bieten, aber ich gebe meine persönliche Meinung (!) ab: Wenn der Instandhalter als Dienstleister des Betreibers den Anwendungscounter zurücksetzt, dann führt das nicht unbedingt zur Rolle des Medizinprodukteherstellers. Das Produkt wird ja (hoffentlich) nicht wieder in Verkehr gebracht, sondern weiter vom gleichen Betreiber benutzt. Der hält sich damit nicht an die vom Hersteller vorgegebene Zweckbestimmung bzw. den bestimmungsgemäßen Gebrauch des Produktes bezüglich der Lebensdauer. Wenn man aber spitzfindig ist, könnte man behaupten, dass der Instandhalter einen Eingriff in die Spezifikation des Produktes macht, denn der Anwendungscounter ist sicher vom Hersteller nicht für den Zugriff vorgesehen. Also hätte der Instandhalter damit das Medizinprodukt so verändert, dass er damit in die Rolle des Herstellers schlüpft und die Verantwortung übernimmt.
Zur Frage „Reicht es also aus, wenn auf Basis einer geeigneten und dokumentierten technischen Prüfung (inklusive Risikomanagement) festgestellt wird, dass das Risiko vertretbar ist und das Produkt weiterhin sicher betrieben werden kann?“:
Der Instandhalter kann niemals eine ausreichende technische Prüfung vorsehen oder Risiken vollständig abschätzen. Das kann nur der Hersteller aufgrund all seiner Daten (auch die der Post-Market-Surveillance). Insofern wäre die uneingeschränkte Haftung beim Instandhalter, der sich überlegen muss, ob er dieses juristische Risiko eingehen will.
Ich empfehle Ihnen hier aber dringend, einen Fachanwalt für Medizinprodukterecht mit einzubinden und sich nicht alleine auf meine Aussage zu stützen.
Herzliche Grüße
Christian Rosenzweig