
Die MEDDEV 2.12-1 ist inzwischen veraltet und wurde durch die MDCG-Leitlinie 2023-3 abgelöst. Eine Zusammenfassung der aktuellen Anforderungen an Vigilanz-Systeme finden Sie in diesem Artikel.
Die MEDDEV 2.12-1 beschreibt die Anforderungen der EU an ein Marktüberwachungs- und Meldesystem (Vigilanz-System). Dieses Vigilanz-System ist Gegenstand von ISO-13485- bzw. Anhang-II-Zertifizierungsaudits.
Regulatorische Relevanz der MEDDEV 2.12-1
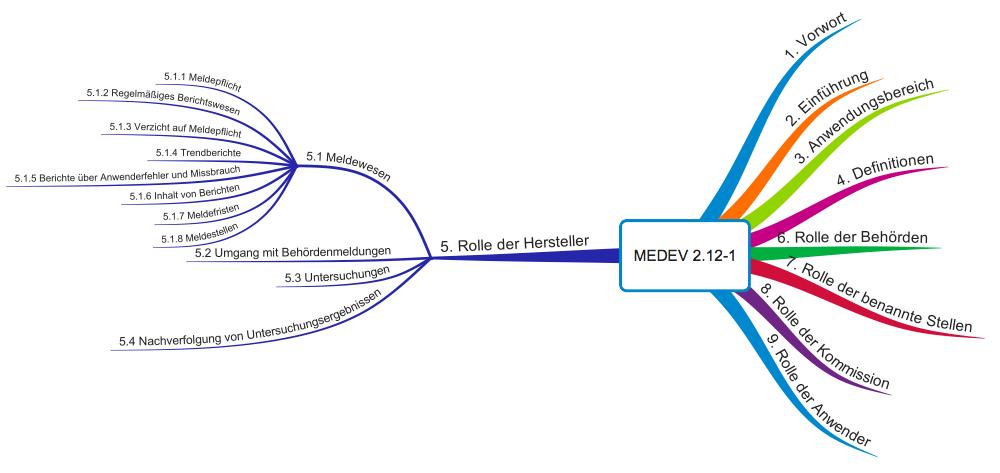
Die MEDDEV 2.12-1 (hier Originaltext) beschreibt nicht nur Anforderungen an die Vigilanz-Systeme von Medizinprodukteherstellern, sondern auch die Rollen
- der Behörden,
- der Benannten Stellen,
- der Europäischen Kommission selbst und
- der Anwender.
Die Hersteller müssen zuerst die nationalen Gesetze und Vorschriften erfüllen. Diese sind in Deutschland mit Bezug zum Meldewesen
- das Medizinproduktegesetz MPG und
- die Medizinproduktesicherheitsplanverordnung MPSV.
a) Forderungen der MEDDEV, die auch die nationalen Gesetze stellen
Diese nationalen Vorschriften sind im Fall von Deutschland an vielen Stellen präziser und verbindlicher als die Forderungen der MEDDEV 2.12-1.
- Meldepflicht (s. u. „Wann gemeldet werden muss“)
- Meldefristen
- Update, z. B. mit Untersuchungsergebnissen und Abschlussbericht
- Inhalt der Meldungen
Die MEDDEV ist dennoch hilfreich, weil sie die Forderungen ausführlicher formuliert und auch Beispiele nennt.
b) Forderungen der MEDDEV, die über die der nationalen Gesetze hinausgehen
Die MEDDEV 2.12-1 stellt auch Forderungen, die über die national-gesetzlichen hinausgehen:
- Trend-Reporting (s. u.)
- Berichte zu Gebrauchstauglichkeitsproblemen und anormalem Gebrauch
- Geräte, mit denen ein Zwischenfall auftrat, habhaft werden
Forderungen der MEDDEV 2.12-1 an das Vigilanz-System der Hersteller
Wann gemeldet werden muss
Wie die MPSV müssen Hersteller einen Zwischenfall nur dann melden, wenn
- ein Fehler am Gerät, eine unbekannte Nebenwirkung, eine inadäquate Therapie oder ein fehlerhaftes Labeling aufgetreten ist UND
- das Gerät für dieses Problem mit-ursächlich sein könnte UND
- der Tod oder eine schwere Gesundheitsbeeinträchtigung eingetreten ist oder hätte eintreten können.
Ob die Informationen über mögliche Probleme direkt von den Anwendern, den Medizinprodukteberatern, den Behörden selbst (z. B. Weiterleiten von Anwendermeldungen an die Behörde) oder aus sonstigen Quellen stammen, ist unerheblich.
Was gemeldet werden muss
Die Meldungen an die Behörden müssen schlussendlich folgende Informationen enthalten:
- Betroffene Geräte, Batch-Nummern
- Problembeschreibung
- Mögliche oder tatsächliche Risiken und Gefährdungen
- Beschreibung von Maßnahmen und deren Begründung
- Repräsentant des Herstellers (in Deutschland Sicherheitsbeauftragter)
Wie schnell gemeldet werden muss
Die MEDDEV 2.12-1 stellt nahezu identische Meldefristen wie die MPSV:
- Unverzüglich
- Bei Bedrohung der öffentlichen Gesundheit
- Bei Todesfällen bzw. schweren Gesundheitsstörungen
- Spätestens nach 30 Tagen: in allen anderen Fällen
Reporting
Trend-Reporting: Die Hersteller sollten es auch dann den Behörden melden, wenn sie Trends von Ereignissen machen, die einzeln nicht einer Meldepflicht unterliegen.
Usability-Reporting: Die Hersteller sollen zudem systematisch Benutzungsfehler und den anormalen Gebrauch auswerten und Berichte auf Verlangen vorweisen können.
Guideline zur Guideline
Im Juli 2019 hat die EU eine neue Guideline zur „alten“ MEDDEV 2.12-1 (aus dem Jahr 2013) veröffentlicht mit dem Ziel,
- Definitionen besser verständlich zu machen,
- einen neuen „Incident Report“ und ein Template für die „Field Safety Notice“ einzuführen,
- die Zusammenarbeit der Behörden untereinander besser zu regeln und
- das Zusammenspiel mit der IMDRF genauer zu beschreiben.
Die Templates und Formulare sind auf der Seite der EU verfügbar, ebenso die MIR Codes, die vom IMDRF stammen. Was sich hinter diesen Codes verbirgt, erfahren Sie auf den Seiten des IMDRF.
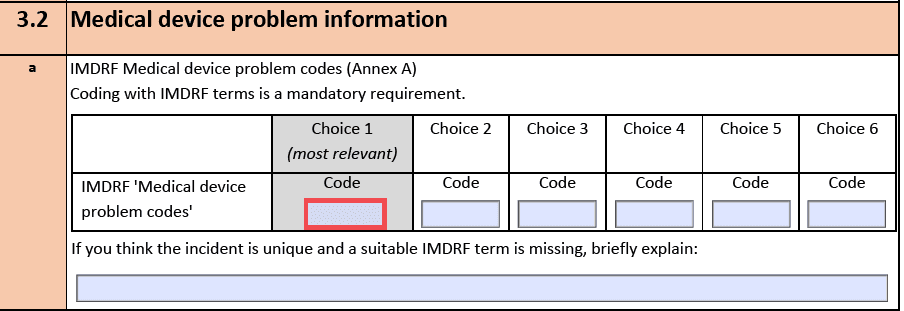
Die Anpassung der Formulare war durch den Umstieg auf die MDR bzw. IVDR notwendig geworden.
Kritik
Kritik an MEDDEV-Dokumenten im Allgemeinen
So hilfreich die MEDDEV-Dokumente sind, so sehr stehen Sie immer wieder in der Kritik. Lesen Sie im Übersichtsartikel zu den MEDDEV-Dokumenten mehr dazu.
Kritik an der MEDDEV 2.12-1 im Speziellen
Bei der MEDDEV 2.12-1 stehen zusätzliche Kritikpunkte im Raum
- Unklare Abgrenzung mit nationalen Vorschriften
Für Hersteller ist es aufwendig herauszufinden, wo die Anforderungen der MEDDEV über die der nationalen Vorschriften hinausgehen. Es sollte auch nicht deren Aufgabe sein. - Fragwürdige Begriffsdefinitionen und Konzepte
Begriffe und Konzepte, wie sie beispielsweise in Normen formuliert sind, finden nur bedingt Berücksichtigung. So führt die MEDDEV 2.12-1 eigene Begrifflichkeiten wie einen indirekten Schaden ein, der mit dem im gleichen Dokument definierten Begriff des Schadens nur schwer in Übereinklang zu bringen ist. - Mangelnde Handlungsleitung
Wesentliche Fragen lässt die MEDDEV unbeantwortet:- Welche Informationsquellen sollen Hersteller auswerten?
- Wie oft sollte das geschehen? Wovon hängt diese Frequenz ab?
- Welche Algorithmen zur Auswertung stehen zur Verfügung?
- Woran kann man erkennen, ob ein Trend meldepflichtig ist?
Bei aller Kritik: Weil sich die MEDDEV 2.12-1 gleichermaßen an Hersteller, Behörden und Benannte Stellen richtet, besteht zumindest die Hoffnung, dass diese Parteien über ein einheitlicheres Verständnis dessen verfügen, was mit Bezug auf ein Vigilanz-System regulatorisch gefordert ist.


Hallo, wie ist die Vorgehensweise bei der Meldung eines Vorkommnisses/eines Sicherheitshinweises gegenüber den nationalen Behörden. Muss der Hersteller tatsächlich alles in die jeweilige Landessprache übersetzen, bzw. die nationalen Formulare in der Landessprache ausfüllen. Falls nein, in welchem Abschnitt ist das beschrieben? Vielen Dank für eine Antwort
Sehr geehrte Frau Wießler,
die Behördenmeldungen erfolgen wie die Registrierung im jeweiligen Land, meist in der jeweiligen Landessprache. Viele Hersteller veröffentlichen aber nur englische Meldungen.
Einen (einzigen) Abschnitt, der dies regelt gibt es nicht, weil die jeweiligen Ländergesetze die Vorgaben machen.
Beste Grüße, Christian Johner
Herzlichen Dank für die umfassende und prompte Antwort
Guten Tag,
ich bin noch recht neu auf dem Gebiet MP und Vigilanz. Und ich frage mich, welcher Zeitrahmen allgemein mit „unverzüglich“ akzeptiert ist.
Auch frage ich mich wie die Definition „Hersteller“ in der EU ist. In den USA ist das ja recht klar „any employee“.
Die 10 Tage Frist würde dann starten, wenn „any employee“ über den Vorfall informiert wird, oder wird das ähnlich gehandhabt wie in den USA?
Herzlichen Dank
Julia W.
Sehr geehrte Julia W,
der Hersteller ist i.d.R. das Unternehmen, keine konkrete Person. Die Definition des Begriffs finden Sie u.a. in der MDR.
Unverzüglich heißt ohne weitere Verzug d.h. sofort. Meist interpretiert man das als „am gleichen Tag“. Wenn es abends ist, würde man auch „am nächsten Tag“ akzeptieren.
Dass der Hersteller seine Mitarbeiter anleiten bzw. Prozess etabliert haben muss, um diese Fristen halten zu können, ist zutreffend.
Mit den besten Grüßen
Christian Johner
Guten Tag,
wie verhält es sich mich der Meldung und Distributoren? Tickt die Uhr von 2 Tagen bei einem Fall mit schwerwiegenden Gefahr für die öffentliche Gesundheit bereits wenn mein Distributor davon erfahren hat, oder erst wenn ich als Hersteller davon Kenntnis habe?! Der Hersteller muss erst mal informiert werden, um aktiv zu werden. Die MDR schreibt nichts dazu wann ein Kunde (Distributor) diese Information weiterleiten muss. Erst wenn der Hersteller informiert wurde tickt die Uhr. Wie sehen Sie das?
Vielen Dank
Dominik S.
Sehr geehrter Herr S.,
die Frist läuft, wenn der Hersteller Kenntnis genommen hat. Meldefristen von Betreibern wird künftig eine nationale Verordnung regeln. Das gibt §88 MPDG in Absatz 7 vor.
Hersteller sollten mit den Distributoren QSVs abschließen, die zur sofortigen Meldungen möglicherweise schwerwiegender Ereignisse verpflichten.
Beachten Sie, dass die MDR heute um ein Jahr verschoben wurde.
Viele Grüße, Christian Johner
Auslegung bzgl. ABC-Kriterien in der MEDDEV 2 12-1
Kriterium „A: An event has occurred“: „Event“ ist gleich „Vorkommnis“, wie in der MDR Artikel 2, Begriffsbestimmungen unter 64 definiert nehm ich an. Wann geht es bei einer Rückmeldung aus dem Feld NICHT um ein Vorkommnis?
Dieses Kriterium ist doch IMMER erfüllt, oder irre ich mich? Beispiele wann KEIN Vorkommnis wären hilfreich.
Lieber Herr Meier,
in der Tat umfasst die Definition von Vorkommnis aus der MDR eigentlich alles, was an negativen Rückmeldungen aus dem Feld zu erwarten ist und somit würde jede negative Rückmeldung diesen Prozess durchlaufen. Was Sie aber vorher ausschließen könnten, sind z.B. positive Rückmeldungen, Rückfragen von Kunden oder auch Kundenwünsche oder Vorschläge bzw. Rückmeldungen ohne konkrete Aussagen zur Produktleistung.
Beste Grüße
Andrea Seeck
Guten Tag,
in wieweit sollte die MEDDEV 2.12-1 noch angewendet werden?
Im Kontext, das dieses Dokument vor der MDR geschrieben wurde und es mittlerweile die „MDCG 2023-3“ (Questions and Answers on vigilance terms and concepts as outlined in the Regulation (EU) 2017/745 on medical devices) vom Februar 2023 gibt?
Viele Grüße
Alexandre Ehlers
Guten Tag Herr Ehlers,
die MEDDEV 2.12-1 sollte unter der MDR nicht mehr angewandt werden. Ein entsprechender Hinweis findet sich im neuen MDCG-Guidance:
Please note that the MEDDEV 2.12/1 rev. 8, January 2013 was in operation under the Directives (Directive 93/42/EEC
concerning medical devices (MDD) and Directive 90/385/EEC on the approximation of the laws of the Member States relating to
active implantable medical devices (AIMDD)) and is not applicable under the MDR.
Viele Grüße
Luca Salvatore
Guten Tag,
im Falle eines schwerwiegenden Vorkommnises: Wem genau muss ich dieses Vorkommnis melden? Zuständige Behörde UND der benannten Stelle? Wie schaut es mit den nicht EU Ländern aus, wenn dasselbe Produkt z.B. eine Zulassung in China oder sogar in Taiwain , Japan und Australien (MDSAP) hat ?
Schöne Grüsse
Jochen Brunner
Guten Tag Herr Brunner,
generell erfolgt die Meldung an die zuständige Behörde, d.h. in dem Land, in dem das Vorkommnis aufgetreten ist. Benannte Stellen sind zu informieren, wenn von dem Produkt eine schwerwiegende Gefahr ausgeht. Im Regelfall möchten die Benannten Stellen immer über schwerwiegende Vorkommnisse informiert werden, was im Vertrag mit dem Hersteller geregelt ist.
Andere Nicht-EU-Länder haben eigene Anforderungen an die Meldung von Vorkommnissen. So müssen z.B. in den USA auch Vorkommnisse gemeldet werden, die außerhalb der USA aufgetreten sind, aber den Meldekriterien in den USA entsprechen.
Sicherheitskorrekturmaßnahmen im Feld (FSCAs), die Sie außerhalb der EU durchführen für ein Produkt, das auch in der EU vermarktet wird, müssen in der EU gemeldet werden.
Herzliche Grüße
Luca Salvatore