
Die Medical Device Coordination Group (MDCG) – auf Deutsch Koordinierungsgruppe Medizinprodukte – ist ein von MDR und IVDR gefordertes Expertengremium.
Die MDCG wird gelegentlich mit einer anderen Koordinierungsgruppe oder mit Expertengremien (Expert Panels) verwechselt. Wie sich diese Gruppen abgrenzen, erfahren Sie hier.
Lesen Sie in diesem Artikel, wie Sie von den Ergebnissen der MDCG-Arbeiten betroffen sind.
1. Wer ist Mitglied der MDCG?
Gemäß MDR ist die MDCG, die Koordinierungsgruppe Medizinprodukte, ein Expertengremium, „das sich aus von den Mitgliedstaaten aufgrund ihrer Rolle und ihres Fachwissens im Bereich Medizinprodukte einschließlich In-vitro-Diagnostika benannten Personen zusammensetzt und das die Kommission berät und die Kommission und die Mitgliedstaaten bei der einheitlichen Durchführung dieser Verordnung unterstützt.“
Nach Artikel 103 MDR werden „die Mitglieder der Koordinierungsgruppe Medizinprodukte […] aufgrund ihrer Fachkompetenz und Erfahrung auf dem Gebiet der Medizinprodukte und der In-vitro-Diagnostika ausgewählt.“ Anderseits macht der Artikel auch klar: „Sie vertreten die zuständigen Behörden der Mitgliedstaaten.“
Die Mitgliedsstaaten entsenden mehrere Mitglieder:
„Jeder Mitgliedstaat ernennt für die Koordinierungsgruppe Medizinprodukte […] ein Mitglied und ein stellvertretendes Mitglied, jeweils mit Fachwissen im Bereich der Medizinprodukte, sowie ein Mitglied und ein stellvertretendes Mitglied mit Fachwissen im Bereich der In-vitro-Diagnostika. […]“. Damit hat die Koordinierungsgruppe ca. 60 Mitglieder.
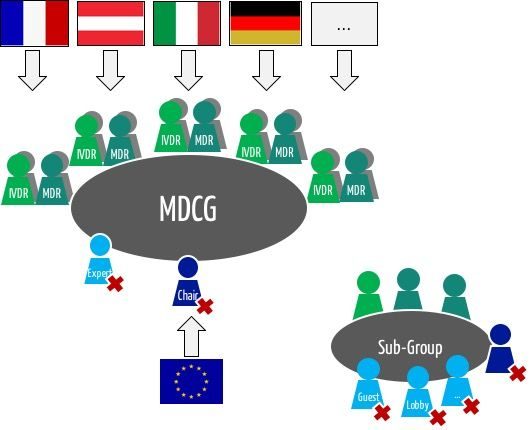
„Die Koordinierungsgruppe Medizinprodukte kann in Einzelfällen Experten und Dritte zur Teilnahme an Sitzungen oder zur Abgabe schriftlicher Beiträge einladen.“
Die MDCG darf Untergruppen einsetzen, um „Zugang zu dem erforderlichen fundierten Fachwissen im Bereich Medizinprodukte einschließlich In-vitro-Diagnostika zu haben“, wie die MDR schreibt. Ob diese Untergruppen sich mit den „Expert Panels“ (siehe unten) überschneiden, ist derzeit noch unklar.
Dreizehn nach Themen geordnete Untergruppen bieten Beratung an und erstellen Leitlinien zu ihren Fachgebieten.
Die Mitglieder der Untergruppen werden von den Mitgliedstaaten für eine Amtszeit von drei Jahren ernannt. Interessenträger nehmen als Beobachter teil. Sie werden nach einem Aufruf zur Einreichung von Bewerbungen ebenfalls für drei Jahre ernannt.
Die Mitglieder der Koordinierungsgruppe tagen regelmäßig unter Vorsitz der EU-Kommission.
2. Aufgaben der MDCG
Die Medical Device Coordination Group (MDCG) hat v. a. beratende und koordinierende Funktionen. Sie ist nicht entscheidungsbefugt und wird von der Kommission nur angehört, bevor sie, die Kommission, beispielsweise
- entscheidet, ob ein Produkt in den Geltungsbereich der Verordnung fällt (Artikel 4, Artikel 51),
- gemeinsame Spezifikationen (Common Specifications) verabschiedet (Artikel 9) oder
- Details zum Meldewesen festlegt (z. B. Form, Fristen, Maßnahmen) (Artikel 91).
Die Kommission hört die MDCG an, wenn sie Expertengremien oder Fachlaboratorien benennt sowie Berater in Expertengremien beruft und in ein zentrales Verzeichnis aufnimmt (Artikel 106).
Die MDCG erstellt ein Marktüberwachungsprogramm, das die zuständigen Behörden beachten müssen (Artikel 93, 105).
Die MDCG kann bei begründeten Bedenken die Expertengremien um wissenschaftliche Gutachten zur Sicherheit und Leistung eines Produkts ersuchen (Artikel 55).
- Beaufsichtigung der Benannten Stellen (NBO)
- Normen
- Klinische Prüfung und Bewertung (CIE)
- Überwachung nach dem Inverkehrbringen und Vigilanz (PMSV)
- Marktüberwachung (MS)
- Borderline-Produkte und Klassifizierung (B&C)
- Neue Technologien
- EUDAMED
- Einmalige Produktkennung (UDI)
- Internationale Angelegenheiten
- In-vitro-Diagnostika (IVD)
- Nomenklatur
- Anhang–XVI–Produkte
a) EUDAMED
Auch im Kontext der EUDAMED muss die Kommission die MDCG anhören (nicht um Zustimmung bitten), bevor sie
- eine UDI-Datenbank (Artikel 28) errichtet oder betreibt,
- ein System, mit dem einmalige Registrierungsnummern generiert werden (Artikel 30), errichtet oder betreibt
- ein System, das Informationen über Benannte Stellen und Konformitätsbescheinigungen verwaltet (Artikel 57), errichtet und betreibt,
- die EUDAMED (Artikel 33) errichtet, unterhält und pflegt sowie
- dazu einen Prüfbericht veröffentlicht (Artikel 34).
b) Benennung Benannter Stellen
Eine wichtige Rolle spielt die Koordinierungsgruppe Medizinprodukte (die MDCG) bei der Benennung und Überprüfung Benannter Stellen.
- Die MDCG beruft gemeinsam mit der Kommission ein Bewertungsteam, das Anträge Benannter Stellen auf Benennung prüft (Artikel 39).
- Die MDCG gibt eine Empfehlung, ob die Benennung erfolgen soll. Ein Abweichen von dieser Empfehlung müssen die (nationalen) Behörden (z. B. ZLG) begründen (Artikel 43).
- Die MDCG wird bei der Überwachung und Neubewertung der Benannten Stellen eingebunden werden (Artikel 44, 45). Das gilt auch, wenn die Kompetenz einer Benannten Stelle angefochten wird (Artikel 46).
3. Veröffentlichungen der MDCG
Die MDCG veröffentlicht ihre Dokumente im DocsRoom der EU.
Neben den Originalveröffentlichungen können wir Ihnen die folgenden (nicht offiziellen) deutschen Übersetzungen bereitstellen:
Die MDCG hat den Zeitplan für die Veröffentlichung weiterer Leitlinien im April 2022 publiziert.
Die neusten Originalveröffentlichungen sind:
Die Veröffentlichungen erschienen vorwiegend zu den Zeitpunkten, an denen die MDR Gültigkeit erlangen sollte bzw. erlangte.
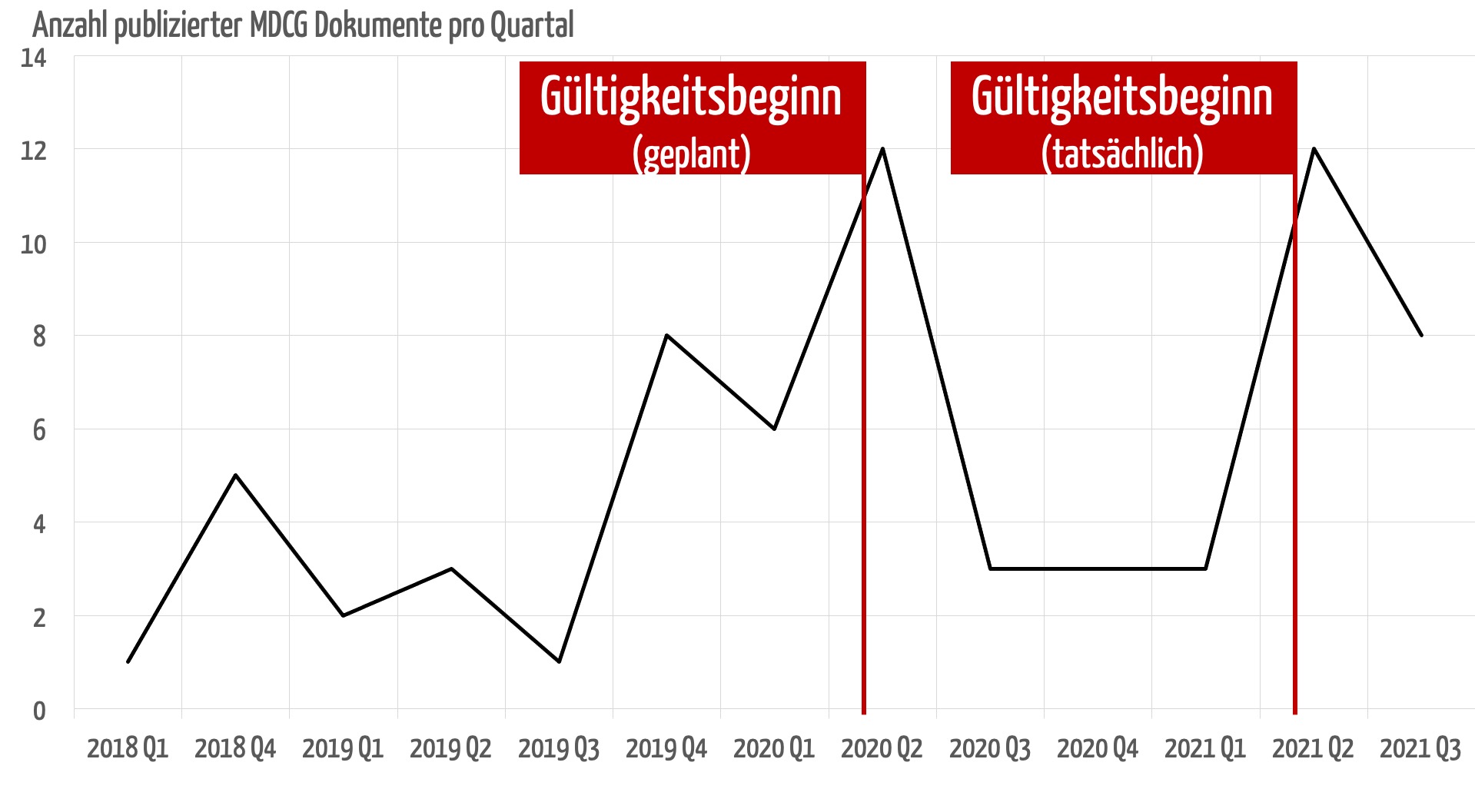
Ein regulatorisches System sollte vollständig und rechtzeitig kommuniziert sein, um von den betroffenen Parteien auch umgesetzt werden zu können. Die MDR erschien im Mai 2017. Zu diesem Zeitpunkt wäre es wünschenswert und hilfreich gewesen, wenn alle MDCG-Leitlinien veröffentlich worden wären. Das ist auch vier Jahre später noch nicht der Fall.
Im Oktober 2019 hat die MDCG eine Roadmap für weitere Dokumente veröffentlicht, die in den nächsten Monaten und Jahren erstellt oder überarbeitet werden sollen.
Sie können hier die Roadmap der MDCG (Stand Oktober 2021) herunterladen.
a) MDCG zu „MDSW under MDR or IVDR“
Mittlerweile veröffentlicht ist das Dokument „Guidance on Classification for Software in MDR 2017/745 and IVDR 2017/746“. Darin beleuchtet die MDCG, wie „Medical Device Software“ (MDSW) unter MDR und IVDR zu klassifizieren und zu bewerten ist. Das Dokument hat den Anspruch, zumindest teilweise die Grundlage für einen Nachfolger der MEDDEV 2.1/6 zu bilden.
i) Software, die ausschließlich beeinflusst, soll keine MPSW sein?
Der Schlussfolgerung der MDCG, dass eine Software, die ausschließlich ein Medizinprodukt kontrolliert und beeinflusst, keine Medical Device Software ist, mag verwirren:
Wenn beispielsweise die Steuerung eines OP-Roboters komplett in eine externe Software ausgelagert ist und diese z. B. basierend auf Input-Signalen entscheidet, ob ein Gewebe krank ist und weggeschnitten werden oder als gesundes Gewebe geschont werden muss, dann spricht das sehr wohl für eine medizinische Zweckbestimmung. Damit müsste eine solche Software ein Medizinprodukt (oder ein Zubehör) sein.
Dem widerspricht das Dokument zwar nicht; allerdings ist die Unterscheidung in Software, die zwar ein Medizinprodukt ist, aber keine Medical-Device-Software (MDSW) ist, und Software, die als Medizinprodukt und als MDSW zählt, nicht unbedingt intuitiv.
Eine adäquate Diskussion, unter welchen Umständen eine Software als Zubehör zu klassifizieren ist, wäre wünschenswert.
ii) Umdeutung von Begriffen
Auch sonst wirkt das Dokument eher verstörend. Beispielsweise will es die Begriffe „Prediction“ und „Prognosis“ nur im Kontext von Diagnosen verstanden wissen. Wo steht dies in der MDR oder der IVDR?
Die Vorhersage eines Krankheitsverlaufs ist keine Diagnose; die Wahrscheinlichkeit, dass ein Patient in der Zukunft an einer Krankheit leiden wird, ebenfalls nicht. Trotzdem ist beides eine Vorhersage bzw. Prognose.
iii) Verwirrung
Sowohl die Beispiele als auch die Formulierungen erreichen das Ziel des Dokuments nicht, nämlich Klarheit darüber zu schaffen, wie im Kontext von MDR und IVDR mit Software zu verfahren ist.
Was ist beispielsweise „Software with built-in electronic controls“? Das Gegenteil, nämlich elektronische Regelungen mit eingebauter Software, scheint es eher zu geben.
Zumindest missverständlich erscheint die Diskussion, ob eine Software ein Medizinprodukt ist:
Eine Firmware in einem Fieberthermometer, die nur die Spannung des Thermistors in Temperatur umrechnet, ist natürlich kein Medizinprodukt (aber das Fieberthermometer ist eines). Daran würde sich auch nichts ändern, wenn die Software die Dosis des passenden Medikaments berechnen würde.
iv) Klassifizierung und Regel 11
Die vielleicht wichtigste Konsequenz dieses Dokuments besteht darin, dass sich die Regel 11 keinesfalls nur auf Stand-alone-Software beschränkt ist. Sie finden hier eine ausführliche Diskussion zur Regel 11 auch im Licht dieser MDCG-Veröffentlichung.
v) Fazit
Es bleibt zu hoffen, dass das Dokument in dieser Form keine Gültigkeit erlangt. Die Hersteller sind mit der mangelnden „Usability“ von MDR und IVDR bereits ausreichend gefordert (lesen Sie beispielsweise Absatz (8) des Artikels 120 der MDR). An Quellen für weitere Verwirrung besteht sicher kein Bedarf; an einer zusätzlichen Verschärfung der Klassifizierung ebenfalls nicht.
b) 2019-16 zur Cybersecurity
Die MDCG möchte nach eigenen Aussagen mit dem 44-seitigen Dokument den Herstellern helfen, die Anforderungen des Anhangs I der MDR bzw. IVDR an die Cybersecurity zu erfüllen. Allerdings beschränkt sie sich weder auf den definierten regulatorischen Rahmen (Anhang I) noch auf die Zielgruppe (die Hersteller).
Relevante Anforderungen von MDR und IVDR
Das Dokument erläutert, welche Anforderungen der EU-Verordnungen insgesamt einen Bezug zur Cybersecurity haben. Diese finden sich zum einen im Anhang I:
- Risikomanagement
- Kombination von Produkten
- Interaktion zwischen Software und IT-Umgebung
- Interoperabilität und Kompatibilität mit anderen Produkten
- Zuverlässigkeit, Wiederholbarkeit, Performanz
- Unautorisierter Zugriff
- Laienanwender
- Begleitmaterialien
Sie zählt aber auch die anderen relevanten Artikel und Anhänge auf:
- Post-Market Surveillance einschließlich Plan und Berichte
- Vigilanz
- Technische Dokumentation
- Klinische Bewertung und Post-Market Clinical Follow-up
Kapitel 2: Basiskonzepte
Das Kapitel über die „Basic Cybersecurity Concepts“ liest sich wie ein Lehrbuch. Es liefert Definitionen, wobei der Begriff Cybersecurity fehlt.
Die Autoren betonen, dass die IT-Sicherheit nur von den Herstellern, Integratoren, Betreibern und Anwendern gemeinsam erreicht werden könne.
Kapitel 3: Entwicklung sicherer Produkte
Ähnlich allgemeingültig bleibt streckenweise auch das dritte Kapitel. Aussagen wie „The primary means of security verification and validation is testing“ sind zwar korrekt, aber von begrenztem Nutzen.
Nützlicher, wenn auch nicht überraschend, sind hingegen Beispiele
- zum physischen Zugriffsschutz auf das Medizinprodukt,
- für Sicherheitsmaßnahmen in der Betriebsumgebung wie Firewalls,
- für Sicherheitsmaßnahmen beim Datenverkehr wie Netzwerksegmentierung und
- zu den Anforderungen an das Patch-Management.
Kapitel 4: Dokumentation und Begleitmaterialien
Sehr ausführlich seziert die Leitlinie 2019-16 den Absatz 23 im Anhang I. Dieser stellt Anforderungen an die Gebrauchsanweisung. Die Leitlinie interpretiert die Anforderungen und nennt Aspekte, die die Dokumentation enthalten muss.
Wie die Autoren auf die jeweiligen Beispiele kommen und mit welcher Begründung sie diese aufnehmen, bleibt unklar. Weshalb bedarf es z. B. „Sufficiently detailed network diagrams for end-users“?
Kapitel 5: Post-Market Surveillance und Vigilanz
Relativ unspezifisch zur IT Security bleibt das fünfte Kapitel. Eine Ausnahme bilden die IMDRF-Codes für meldepflichtige Probleme.
Kapitel 6: Verweis auf andere Regularien
Das MDCG 2019-16 Dokument verweist auf weitere regulatorisch relevante Dokumente:
- NIS Richtlinie 2016/1148 (Directive on security of network and information systems)
- DSGVO
- IMDRF Guide on Cybersecurity of Medical Devices
Kapitel 7 bis 10: Anhänge
Das Kapitel 7 enthält einen nützlichen Anhang mit einem Mapping der MDR-Anforderungen auf die Anforderungen der NIS-Richtlinie.
Kapitel 8 nennt Beispiele für IT-Sicherheitsprobleme (Security) und die Auswirkungen auf die Safety.
Kapitel 9 listet relevante Normen.
Kapitel 10 zeigt das Zusammenspiel zwischen dem „normalen“ Risikomanagement und dem „Cybersecurity Risk Management“.
Fazit
Das Dokument lässt einen auch nach mehrfachem Lesen etwas ratlos und mit einem „So what?“ zurück. Wer sich mit der IT-Sicherheit ein wenig auskennt, wird wenig Neues erfahren. Wer das nicht tut, wird das Geschriebene nicht verstehen. Dazu reicht das didaktische Niveau nicht aus.
Es bleibt der Eindruck, dass lediglich ein weiteres Dokument erzeugt wurde, das Grundlagen zur IT-Sicherheit zu erläutern versucht. Dem Anspruch, eine konkrete Handlungsleitung zu geben, wie man die Anforderungen von MDR und IVDR an die IT-Sicherheit erfüllt, wird die Leitlinie nur bedingt gerecht.
Es wäre zu wünschen, dass die MDCG beim Verfassen neuer Dokumente eine ähnliche Stringenz und Methodik bei der Qualitätssicherung übernimmt wie das in (vielen) Normengremien und bei wissenschaftlichen Publikationen üblich ist.
Hersteller benötigen nicht eine Flut an Dokumenten, sondern konkrete Hinweise, wie sie die EU-Verordnungen interpretieren und umsetzen sollen.
4. Abgrenzung zu anderen Gruppen und Gremien
Verwechseln Sie die Koordinierungsgruppe Medizinprodukte (die Medical Device Coordination Group MDCG) nicht mit der Koordinierungsgruppe Benannter Stellen und anderen Expertengremien.
a) Expertengremien / Expert Panels
Die Expertengremien bestehen gemäß Artikel 106 „aus Beratern, die die Kommission auf der Grundlage aktuellen klinischen, wissenschaftlichen oder technischen Fachwissens auf dem betreffenden Gebiet und nach einer geografischen Verteilung berufen hat, die die Vielfalt der wissenschaftlichen und klinischen Konzepte in der Union widerspiegelt. […] Die Mitglieder der Expertengremien erfüllen ihre Aufgaben unparteiisch und objektiv. Sie dürfen Weisungen von Benannten Stellen oder Herstellern weder einholen noch entgegennehmen.“
Diese Expertengremien haben folgende Aufgaben:
- Sie beraten Benannte Stellen und Hersteller „unter anderem im Hinblick auf die Kriterien für einen angemessenen Datensatz für die Konformitätsbewertung eines Produkts, insbesondere in Bezug auf die für die klinische Bewertung erforderlichen klinischen Daten, in Bezug auf die physikalisch-chemische Charakterisierung und in Bezug auf die mikrobiologische, die Biokompatibilitäts-, die mechanische, elektrische, elektronische und nichtklinische toxikologische Untersuchung.“
- Sie erstellen „Gutachten zu den Berichten der Benannten Stellen über die Begutachtung der klinischen Bewertung bei bestimmten mit einem hohen Risiko behafteten implantierbaren Produkten“. Diese Gutachten bzw. Bewertungen sind bei implantierbaren Produkten der Klasse III und aktiven Produkten der Klasse IIb verpflichtend. Die Koordinierungsgruppe Medizinprodukte kann bei „begründeten Bedenken die[se] Expertengremien um wissenschaftliche Gutachten zur Sicherheit und Leistung eines Produkts ersuchen“.
- Zudem unterstützen die Expertengremien die Kommission bei der Erstellung von gemeinsamen Spezifikationen (Common Specifications) insbesondere im Kontext klinischer Bewertungen und klinischer Prüfungen.
- Die Leitlinien wie z. B. die 2019-16 zur Cybersecurity formuliert sehr konkrete Vorgaben, die die Hersteller bei der Entwicklung und Überwachung ihrer Produkte beachten müssen.
b) Koordinierungsgruppe der Benannten Stellen
Die Koordinierungsgruppe Medizinprodukte darf auch nicht verwechselt werden mit der Koordinierungsgruppe der Benannten Stellen, die der Artikel 49 („Koordinierung der Benannten Stellen“) fordert:
„Die Kommission stellt sicher, dass eine angemessene Koordinierung und Zusammenarbeit der Benannten Stellen stattfindet, und zwar in Form einer Koordinierungsgruppe für Benannte Stellen auf dem Gebiet der Medizinprodukte einschließlich In-vitro-Diagnostika. Die Gruppe tritt regelmäßig, jedoch mindestens jährlich zusammen. […] Die Kommission kann die Einzelheiten für die Arbeitsweise der Koordinierungsgruppe für Benannte Stellen festlegen.“
Eine solche Koordinierungsgruppe Benannter Stellen gibt es bereits in Form der Notified Bodies Operation Group NGOB, das mit seinen NBOG-Dokumenten Bekanntheit erreicht hat.
Weiter gibt es das Team-NB mit seinen NB-MED-Dokumenten.
5. MDCG: Zusammenfassung, Fazit und Kritik
a) Wen die MDCG betrifft
Die Arbeit der Medical Device Coordination Group betrifft die Benannten Stellen direkt, die Medizinproduktehersteller und deren Dienstleister ebenso:
- Die MDCG empfiehlt, wann Produkte unter die MDR bzw. IVDR fallen sollen.
- Sie erhöht den Druck auf die Benannten Stellen, der im Audit und bei der Überprüfung der technischen Dokumentation an Sie weitergereicht wird.
- Weiter wird die MDCG angehört, wenn neue Common Specifications erstellt werden, die Sie zwingend erfüllen müssen.
- Hat eine Koordinierungsgruppe Zweifel an der Sicherheit Ihres Produkts, kann sie ein Gutachten bei den Expertengremien anfordern.
b) Unklare Verbindlichkeit
Die MDCG scheint ein weitgehend zahnloser Tiger zu sein. Man überträgt diesem Gremium zwar Arbeit und hört es an. Entscheiden darf es aber nicht.
Allerdings betrachten viele Auditoren Benannter Stellen die Veröffentlichungen der MDCG als „Stand der Technik“ und damit wie Gesetze. Und das, obwohl die MDCG in Ihren Leitlinien selbst schreibt:
The document is not a European Commission document and it cannot be regarded as reflecting the official position of the European Commission. Any views expressed in this document are not legally binding and only the Court of Justice of the European Union can give binding interpretations of Union law.
z.B. MDCG 2019-16
Mit diesem „Disclaimern“ macht es sich die MDCG deutlich zu einfach. Selbst das EuGH hat sich bei seinem Urteil über Software-Komponenten auf das Vorgänger-Dokument der Leitlinie MDCG 2019-11, die MEDDEV 2.1/6, bezogen.
Es muss den Autoren klar sein, dass ihre Veröffentlichungen als bindend verstanden werden.
c) Intransparente und undemokratische Entscheidungen
Gerade weil die Dokumente diesen verbindlichen Charakter haben, kann und darf es nicht sein, dass deren Entstehen völlig intransparent verläuft. Die Tagungen finden hinter verschlossenen Türen statt.
Selbst Mitglieder dieses Gremiums beklagen sich darüber, dass Entscheidungen und Dokumente des Gremiums im Nachgang verändert werden.
Dass Dokumente, die de facto gesetzlichen Charakter haben, vorbei an jedem parlamentarischen Prozess entstehen, ist einer Demokratie nicht würdig. Genau diese Verhaltensweise schürt die Politikverdrossenheit und die Ablehnung der Brüsseler Institutionen.
d) Unklare Kompetenz der „Experten“
Ob die Mitgliedsstaaten wirklich die besten Experten und nicht einfach nur Behördenvertreter in die Koordinationsgruppe berufen, wird sich zeigen. Fast wie ein Eingeständnis liest sich daher die Aussage:
„Die Koordinierungsgruppe Medizinprodukte kann in Einzelfällen Experten und Dritte zur Teilnahme an Sitzungen oder zur Abgabe schriftlicher Beiträge einladen.“
Die bisherigen Veröffentlichungen lassen, wie oben dargelegt, nicht immer den zwingenden Schluss zu, dass diejenigen Personen beteiligt sind, die sich in der jeweiligen Domäne am besten auskennen.
e) Hoffnung
Berechtigter sind hingegen die Hoffnungen, dass die Medical Device Coordination Group
- dazu beiträgt, dass die Qualität der Benannten Stellen europaweit einheitlicher wird,
- die Arbeit der Hersteller nicht noch weiter wesentlich behindert wird und
- freier von finanziellen oder sonstigen Interessen ist, als das Experten aus der Medizinprodukteindustrie oder Lobbyverbänden wären.
Dass diese Hoffnungen gerechtfertigt sind, dürfte angesichts des oben beschriebenen Entwurfs zur Klassifizierung von „Medical Device Software“ bezweifelt werden.
Die MDR bleibt auch im Kontext der „Koordinationsgruppe Medizinprodukte“ dem Grundsatz treu, dass die EU-Kommission überall das letzte Wort hat.
Änderungshistorie
- 2023-05-16: Aktuelle MDCG-Dokumente ergänzt
- 2022-11-25: Aktuelle MDCG-Dokumente ergänzt
- 2022-09-08: Deutsche Übersetzungen von MDCG 2022-14 unter 3. ergänzt
- 2022-05-06: MDCG 2022-6 in der Tabelle ergänzt
- 2022-04-28: MDCG 2022-5 in der Tabelle ergänzt
- 2022-04-11: Zeitplanung der MDCG in Kapitel 3 ergänzt
- 2022-02-25: Aktuelle MDCG-Guidances ergänzt
- 2022-02-11: Deutsche Übersetzungen von Dr. Castner von MDCG-Guidances verlinkt (3.)
- 2022-01-27: Aktuelle MDCG-Guidances ergänzt
- 2021-12-10: Aktuelle MDCG-Dokumente ergänzt
- 2021-11-15: Die Roadmap der MDCG (Stand Oktober 2021) ergänzt
- 2021-07-27: Im 3. Abschnitt die Grafik ergänzt



Lieber Herr Johner,
ihre Aussage „Der Schlussfolgerung der MDCG, dass eine Software, die ausschließlich ein Medizinprodukt kontrolliert und beeinflusst, kein Medizinprodukt sei, kann das Johner Institut nicht folgen“
ist nicht korrekt. Die MDCG Guideline sagt nicht, dass es dann kein Medizinprodukt ist, sondern nicht der Definition einer MDSW entspricht. Es bleibt dann trotzdem ein Medizinprodukt, aber halt keine MDSW. MDSW beschreibt Software, die einen eigenständigen medizinischen Zweck verfolgt (unabhängig davon ob Sie bei Inverkehrbringung „integraler“ Bestandteil ist oder nicht) und deshalb auch optional einem eigenständigen Konformitätsbewertungsverfahren unterzogen werden kann.
Bei den anderen Punkten bin ich bei Ihnen, da ist noch Luft nach oben. Noch sind wir im (finalen) Entwurf und man kann vielleicht das eine oder andere anpassen.
Danke, lieber Herr Hilgers!
Ich wusste nicht, dass Sie Teil des Teams sind. Den Punkt mit dem Zubehör fixe ich sofort.
Nochmals herzlichen Dank für Ihre Unterstützung, dank derer ich den Beitrag weiter verbessern kann.
Viele Grüße, Christian Johner
Lieber Herr Johner,
sehr gerne, ich finde es super, dass Sie die Diskussionen zu diesem wichtigen Thema hier anregen.
Sie erwähnen „Zubehör“, was aber auch nicht so gemeint war. Das Beispiel der Firmware im Fieberthermometer trifft es ganz gut. Diese Firmware wäre keine MDSW weil Sie nur den Zweck des Thermometers unterstützt. Sie wäre dann aber auch kein Zubehör sondern einfach nur Bestandteil des Medizinprodukts „Fieberthermometers“. Hätten wir aber ein Fieberthermometer, dass mit einer Cloud verbunden ist und die Ergebnisse an einen Arzt sendet und einen Alarm verschickt bei Überschreitung von Grenzwerten, würde die Software mehr als nur messen und hätte einen eigenständigen Zweck (alarmieren) und würde dann als MDSW eingestuft werden. Jetzt könnte der Hersteller bei einer gut zerlegten Architektur überlegen die MDSW eigenständig als Produkt abzugrenzen oder weiterhin als ein Gesamtprodukt einem Conformity Assessment zu unterziehen. Die Entscheidung sollte dann in der Regel davon abhängen ob sich die Klassifizierung signifikant unterscheidet. Ich hoffe das hilft weiter zur Klärung.
Sehr geehrter Herr Prof. Johner,
ich wollte an dieser Stelle eine Information einbringen, die als Ergänzung des zum Punkt Klassifizierung/Regel 19/Nanomaterial verwendet werden kann:
Wir hatten firmenintern die Debatte, ob Baumwolle als Nanomaterial zählt, da die Microfibrillen einer Baumwollfaser Dimensionen im Nanobereich aufweisen, und deshalb alle Produkte, die Baumwolle enthalten, unter die Regel 19 fallen würden.
Nach jetzigem Stand scheint Baumwolle wohl tatsächlich unter diese Definition zu fallen, es soll jedoch bis Ende dieses Jahres, Anfang nächsten Jahres ein MDCG-Guidance Document veröffentlicht werden, in der klar gestellt wird, dass das so nie beabsichtigt war.
Vielleicht kann dazu ein MDCG-Mitglied nähere Informationen liefern, meine Informationen sind aus 2. Hand.
Mit den besten Grüßen
Martin von Rüden
Sehr geehrter Herr von Rüden,
vielen Dank für diese Information! Ich schaue mal, ob ich über meine Kanäle etwas rausfinde.
In der Roadmap der MDCG konnte ich das Thema nicht finden. Diese Roadmap wurde gerade erst publiziert. Es kann aber sein, dass es in anderen Guidance Dokumenten diskutiert wir wie dem zur „Classification of Medical Devices“. Dieses Guidance Dokument hat aber noch kein geplantes Datum. Für andere Dokumente gibt es diese Abschätzungen, die sich aber alle auf die Jahre 2019 und 2020 beziehen.
Nochmals besten Dank!
Herzliche Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
gut dass sie die Intransparenz und undemokratischen Entscheidungen der MDCG´s ansprechen. Wenn wir schon in einer Demokratie leben, sollte unser Leben auch demokratisch gestalltet werden. Solche Auswüchse wie Intransparenz und undemokratische Entscheidungen sollte wir auf keinen Fall dulden. Es ist schon schlimm genug, wenn unsere Behöden undemokratisch entscheiden. Dort herrscht Diktatur in Reinstform. Jeder kleine Beamte, der nicht spurt, wird vom Dienst suspendiert oder entlassen. Es kann nicht sein, dass wir „angeblich“ in einer Demokratie leben, die Behörden aber innerhalb ihres Bereiches eine Diktatur pflegen. Wenn die unteren Beamten sich demokratisch einig sind, sollte sie entscheiden und sich nicht von irgendwelchen Vorgesetzten, die aus was für Gründen auch immer eingesetzt wurden, diktatorisch gängeln lassen. Die angebliche Loyalität zum Staat wird hier zur Dikatur. Genauso sollte es mit den MDCG`s gemacht werden. Entweder entscheiden die Mitglieder demokratisch, oder man löst diesen undemokratischen Haufen auf und etabliert eine demokratische Lösung.
Auf der einen Seite schreit man in Deutschland und Europa nach Diversität in Meinung und im Leben, und auf der anderen Seite wird diktatorisch beschlossen und möglichst alles uniform umgesetzt.
Was soll dieses Kasperle-Theater?
Mit freundlichen Grüßen
Wolfgang Willmann
Sehr geehrter Herr Dr. Willmann,
danke für Ihre Gedanken!
Ich hoffe, dass Ihre Kritik nicht mir gilt. Ich hoffte, nur zu berichten.
Beste Grüße, Christian Johner
Sehr geehrte Damen und Herren,
ich wollte nur darauf aufmerksam machen, das einige Links zu MDCG-Dokumente (PDFs) nicht mehr funktionieren.
Aufgefallen ist mir dies ab dem Link zur MDCG 2021-13 Rev. 1 und nachfolgenden älteren Dokumenten. (Bereich: 3. Veröffentlichungen der MDCG)
Viele Grüße
N. Jacobs
Herzlichen Dank, lieber Herr Jacobs!
Wir schauen uns das gleich an. Dank Ihres Hinweises können wir die Links up-to-date bringen.
Nochmals besten Dank!
Viele Grüße
Christian Johner
Wir sind gerade dabei Produkte in die Eudamed Datenbank zu registrieren.
Bei den EMDN Codes gibt es grobe Möglichkeiten eine Möglichkeit zu dokumentieren, aber die Codes passen nicht wirklich. Wir müssten in einer besser passenderen Kategorie einen neuen Code beantragen.
Wie läuft die Beantragung von Codes?
Mit freundlichen Grüßen
D. Gutzler
Sehr geehrter Herr Gutzler,
ich bin unsicher, ob es vorgesehen ist, dass neue Codes beantragt werden können. Ein entsprechendes Vorgehen ist mir leider nicht bekannt. Meine Empfehlung wäre, eine Anfrage an die verantwortliche Person seitens der Europäische Kommission zu stellen.
Viele Grüße
Sehr geehrter Herr Prof. Johner,
haben alle MEDDEV Dokumente mittlerweile ihre Wirksamkeit verloren und wurde diese durch MDCG Leitlinien ersetzt? Wir haben diesbezüglich bei Audits (Produkte MDD Übergangsphase MDR) von unterschiedlichen benannten Stellen widersprüchliche Aussagen. Gibt es hierzu ein verbindliches Wording?
Besten Dank vorab,
Astrid Unterkreuter
Sehr geehrte Frau Unterkreuter,
leider besteht eine rechtliche Unklarheit bzgl. der Verbindlichkeit der MEDDEV und auch MDCG Dokumente. Viele (jedoch nicht alle) Auditoren von Benannten Stellen erachten diese Dokumente jedoch als „Stand der Technik“, weshalb diese als Argumentation genutzt werden können. (Selbst das EuGH hat sich bei seinem Urteil über Software-Komponenten auf das Vorgänger-Dokument der Leitlinie MDCG 2019-11, die MEDDEV 2.1/6, bezogen.)
Aufgrund dieser Situation kann ich Ihnen leider kein „verbindliches Wording“ mitteilen.
Herzliche Grüße
Sehr geehrter Herr Gerhart,
bezüglich der neuen MDCG 2023-3 wird unter der Note 1 (Seite 1/18) offiziell mitgeteilt, dass
„[…] the MEDDEV 2.12/1 rev. 8, January 2013 was in operation under the Directives […] and is not applicable under the MDR.“. Für unsere Vorkommnis Bewertungen haben wir die MEDDEV bisher angezogen, da uns diese Guideline die Möglichkeit bot, unter der Betrachtung des Kapitels 5.1.3.6 gewisse Vorkommnisse nicht melden zu müssen.
Wenn nun die MEDDEV 2.12/1 rev.8 keine Anwendung mehr findet, wo finde ich nun Informationen zum FSCA , Trend sowie dem Periodic Summary Report Template?
Können Sie mir bestätigen, dass mit dem MDCG 2023-3 die MEDDEV 2.12/1 rev. 8 nicht mehr anwendbar ist?!
Herzlichen Dank im Voraus.
Viele Grüße
C.Dittmar
Sehr geehrter Herr Dittmar,
das neue MDCG 2023-3 Dokument definiert, dass das MEDDEV 2.12/1 rev. 8 Dokument nicht für die MDR anwendbar ist. Mir ist leider kein anwendbares vergleichbares Dokument bekannt, abgesehen von dem Dokument MDCG 2023-3. Jedoch können Sie Teile (bspw. Templates) aus dem Dokument MEDDEV 2.12/1 rev. 8 als Hilfestellung nutzen, wobei Sie vorab prüfen müssen, ob die Templates den ggf. definierten Anforderungen in der MDR entsprechen bzw. diese erfüllen.
Herzliche Grüße
Guten Tag,
ist Ihnen bekannt, ob und wie es möglich ist gewisse Themen, für die ein Guidance-Dokument wünschenswert wäre, bei der MDCG einzusteuern?
Es gibt trotz der fortschreitenden Zeit immer noch Themenbereiche, die bisher komplett ignoriert worden und nicht in den Plänen für weitere Guidances berücksichtigt sind.
Vielen Dank vorab.
MfG
C. Bausch
Sehr geehrte Frau Bausch,
sie können über den Link prüfen, ob Ihr Thema ggf. bereits in Bearbeitung ist. Sollte dies nicht der Fall sein, könnten Sie sich an die themenspezifischen Arbeitsgruppenleiter unter dem Link wenden.
Herzliche Grüße